
什么是GSEA富集分析
当我们设置好分组实验最后获得各组对基因的表达矩阵时,我们通常希望得到不同分组中具有差异表达的基因。
如果进行常规差异分析,通过log2fc筛选差异基因,就是将筛选差异基因的标准聚集在单个基因的差异表达上。
而实际上每个基因的差异表达造成的表型差异有所不同,有些基因虽然差异表达较小,但会造成较大的生理功能变化。并且生理功能通常是由一系列基因调控的,所以只将筛选标准聚集在单个基因差异表达上,可能会错过真正具有生物学含义的基因群差异。
于是我们可以选择进行GSEA富集分析,该种分析聚集于基因群的差异,需要一个预设的基因集S(我们通常使用一个通路中的所有基因作为基因集S),而我们实验所获得的所有基因集L,则通过某种标准的排序后(通常按表达差异倍数log2fc排序),作为通路基因集S的打分标准。
对于一个通路的ES打分步骤为:从基因集L的第一个基因开始到最后一个基因进行判断,如果该基因属于基因集S,则加分,不属于基因集S则减分(得分的大小由其相关性决定)。
如果我们将基因集S在L的每一个位置的ES得分点实时标注出来,就会得到一条曲线,该条曲线的斜率表示了L基因集在通路基因集S中的富集程度,斜率越大富集程度越高,所以与该通路中最相关的基因就集中在斜率最大那段的曲线上。
如果我们的基因集L是按照log2fc排序的,就可预见如我们获得的基因中存在与该通路正相关的基因集,那么会在y轴上方具有一个峰,该峰左边即为S通路的富集基因集;若我们获得基因中存在与该通路负相关的基因集,那么会在y轴下方具有一个峰,该峰右边即为S通路的富集基因集。
当然,我们可以自由构建不同的基因集L,S,如S换成我们感兴趣的基因集,L按其他标准排序都是可以的,通过斜率就可以直观地得到L关于S的富集基因集。
GSEA富集分析需要哪些数据
1.表达矩阵所有基因的ENTREZID编号
2.所有基因差异分析所得log2FoldChange
3.以上组成一个数据框,按log2FoldChange升序排列
4.取出log2FoldChange作为向量,将其对应的ENTREZID编号作为其names
rm(list = ls()) options(stringsAsFactors = F) load("./Rdata/ENTREZID.Rdata") library(org.Mm.eg.db) library(clusterProfiler) library(enrichplot) library(tidyverse) library(ggstatsplot) library(GseaVis) data<-deg[,c(2,9)] data<-data[order(data$log2FoldChange,decreasing = T),] geneList <- data$log2FoldChange names(geneList) <- data$ENTREZID讯享网
GSEA富集分析代码
除clusterProfiler包外,还有其他可以GSEA分析的方法
讯享网GSEA富集分析代码 除clusterProfiler包外,还有其他可以GSEA分析的方法 organism = 'mmu' # 人类'hsa' 小鼠'mmu' OrgDb = 'org.Mm.eg.db' KEGG_kk_entrez <- gseKEGG(geneList = geneList, organism = organism, #人hsa 鼠mmu pvalueCutoff = 1) #实际为padj阈值可调整 KEGG_kk <- DOSE::setReadable(KEGG_kk_entrez, OrgDb=OrgDb, keyType='ENTREZID')#转化id GO_kk_entrez <- gseGO(geneList = geneList, ont = "ALL", # "BP"、"MF"和"CC"或"ALL" OrgDb = OrgDb,#人类org.Hs.eg.db 鼠org.Mm.eg.db keyType = "ENTREZID", pvalueCutoff = 1) #实际为padj阈值可调整 GO_kk <- DOSE::setReadable(GO_kk_entrez, OrgDb=OrgDb, keyType='ENTREZID')#转化id save(KEGG_kk_entrez,KEGG_kk,GO_kk_entrez,GO_kk, file = "./Rdata/GSEA_result.RData") kk_gse <- KEGG_kk kk_gse_entrez <- KEGG_kk_entrez kk_gse_cut <- kk_gse[,kk_gse$pvalue<1 & kk_gse$p.adjust<1 & abs(kk_gse$NES)>1] kk_gse_cut_down <- kk_gse_cut[kk_gse_cut$NES < 0,] kk_gse_cut_up <- kk_gse_cut[kk_gse_cut$NES > 0,]
GSEA富集分析可视化
1.gseaplot2/enrichplot+ggplot2:可以通过ggplot2加上所需的标注,自由度高
2.GseaVis包的gseaNb函数:给的参数较多,调整较为简便

具体参数推荐参考文章:
高分文章呼唤高颜值的GSEA可视化方式 - 知乎 (zhihu.com)
下载代码:devtools::install_github("junjunlab/GseaVis")
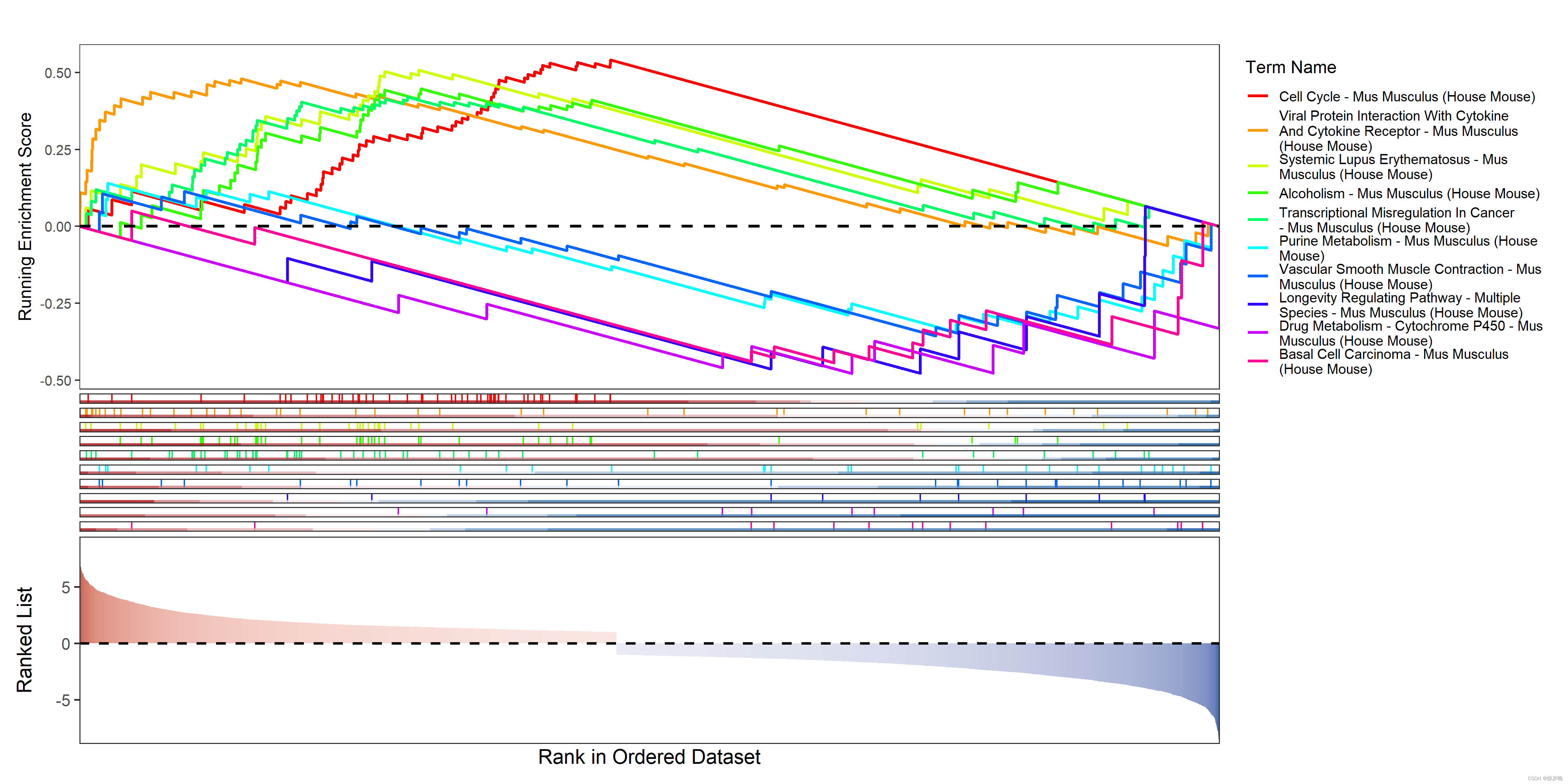
讯享网#选择展现NES前几个通路 down_gsea <- kk_gse_cut_down[tail(order(kk_gse_cut_down$NES,decreasing = T),10),]#负调控 up_gsea <- kk_gse_cut_up[head(order(kk_gse_cut_up$NES,decreasing = T),10),]#正调控 gseap1<-gseaNb(object = kk_gse, geneSetID = c(row.names(up_gsea)[1],row.names(down_gsea)[1]),#绘制哪几个通路的曲线 subPlot = 3,#保留几个副图 addPval = T,#是否显示pval表格 pvalX = 1,pvalY = 2,#pval表格位置 curveCol=rainbow(10)#自动提供3种颜色,超过四个通路需要手动设置颜色 ) gseap1 width=15 height=7.5 ggsave(plot = gseap1,filename = './PDF/gseap1.pdf', width = width,height = height) ggsave(plot = gseap1,filename = './PNG/gseap1.png', width = width,height = height) gseap2 <- gseaNb(object = kk_gse, geneSetID = c(row.names(up_gsea)[c(1:5)],row.names(down_gsea)[c(1:5)]),#绘制哪几个通路的曲线 subPlot = 3,#保留几个副图 #termWidth = 35,#调整图例位置和长度 #legend.position = c(0.8,0.3),#调整图例位置和长度 addPval = F,#是否显示pval表格 pvalX = 0.05,pvalY = 0.05,#pval表格位置 curveCol=rainbow(10)#自动提供3种颜色,超过四个通路需要手动设置颜色 ) gseap2 width=15 height=7.5 ggsave(plot = gseap2,filename = './PDF/gseap2.pdf', width = width,height = height) ggsave(plot = gseap2,filename = './PNG/gseap2.png', width = width,height = height)
结果
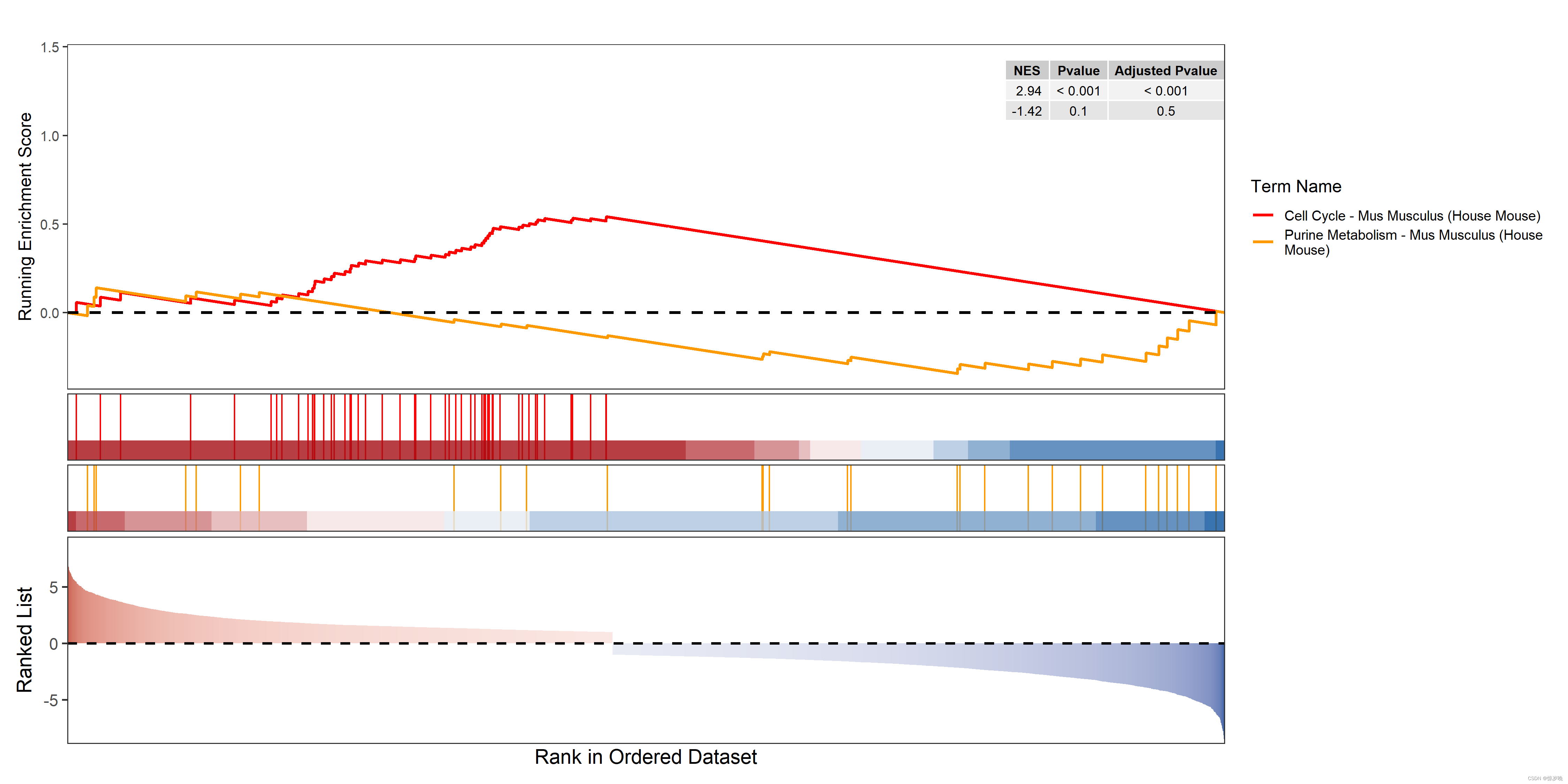
gseap1:
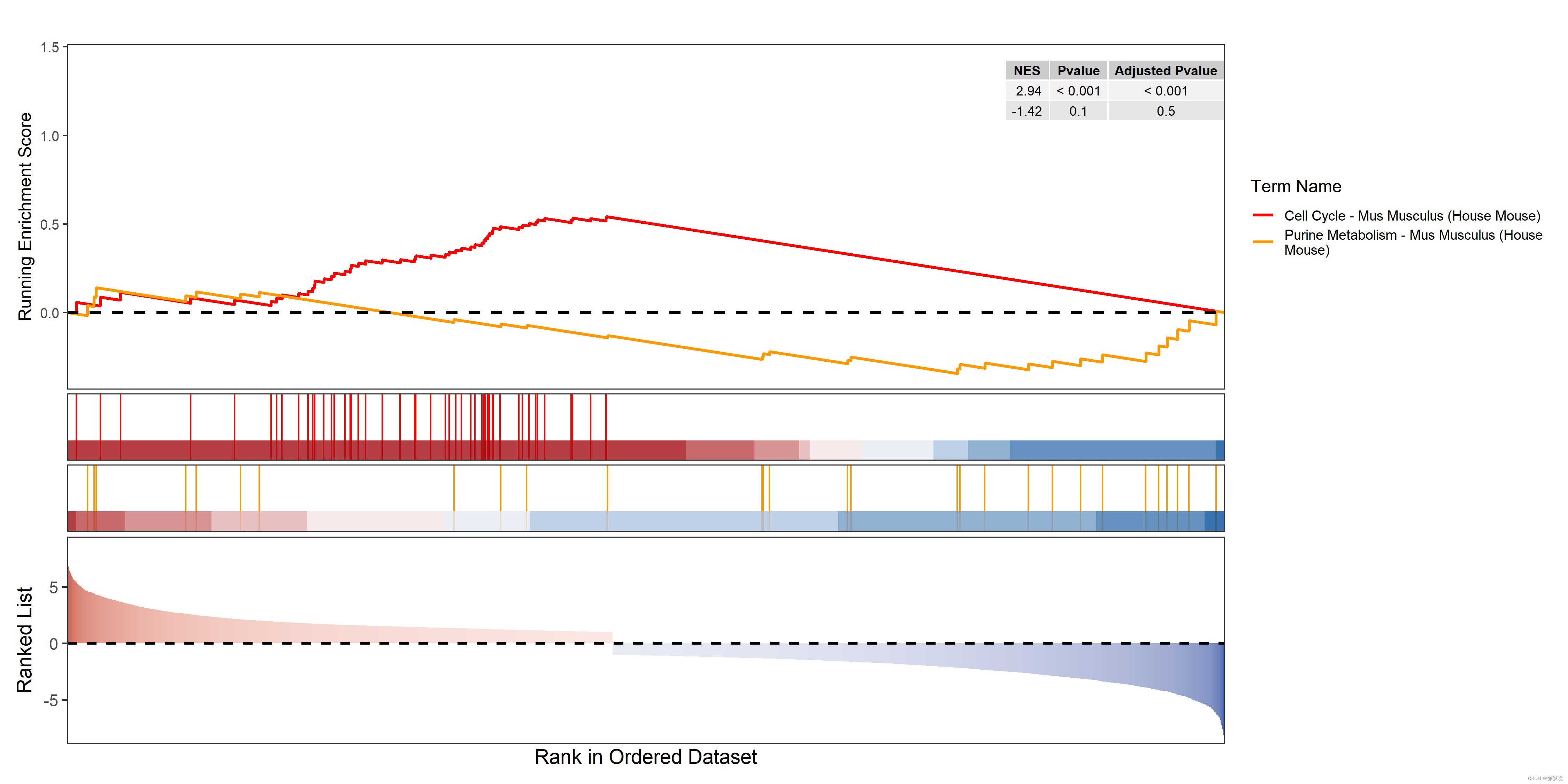
gseap2:
获取核心基因
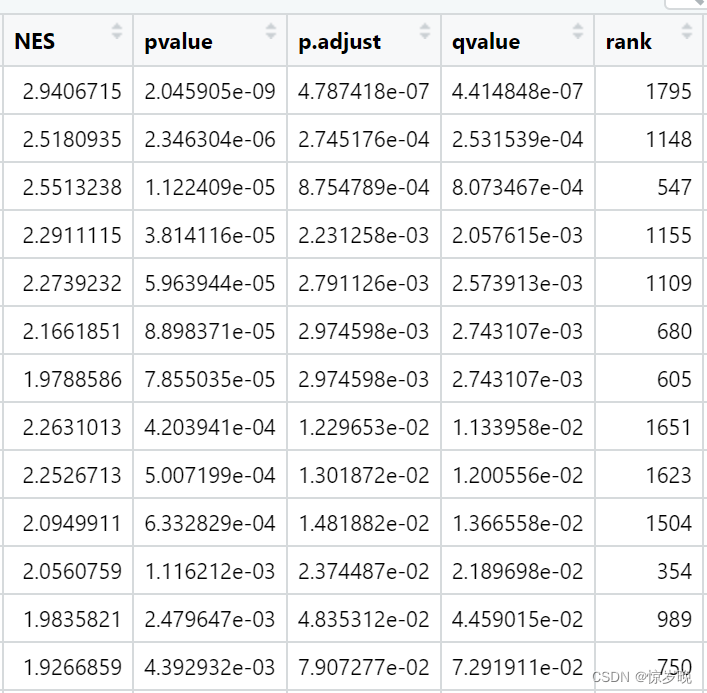
rank为顶点基因在L的排序,取其附近基因即为核心基因
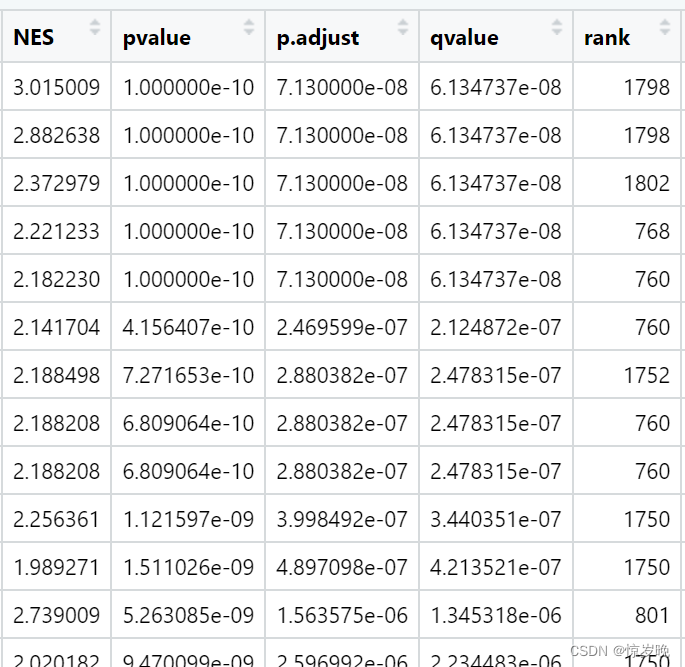
KEGG数据库比对的结果:
GO数据库比对的结果:
整体代码
rm(list = ls()) options(stringsAsFactors = F) load("./Rdata/ENTREZID.Rdata") library(org.Mm.eg.db) library(clusterProfiler) library(enrichplot) library(tidyverse) library(ggstatsplot) library(GseaVis) data<-deg[,c(2,9)] data<-data[order(data$log2FoldChange,decreasing = T),] geneList <- data$log2FoldChange names(geneList) <- data$ENTREZID organism = 'mmu' # 人类'hsa' 小鼠'mmu' OrgDb = 'org.Mm.eg.db' KEGG_kk_entrez <- gseKEGG(geneList = geneList, organism = organism, #人hsa 鼠mmu pvalueCutoff = 1) #实际为padj阈值可调整 KEGG_kk <- DOSE::setReadable(KEGG_kk_entrez, OrgDb=OrgDb, keyType='ENTREZID')#转化id GO_kk_entrez <- gseGO(geneList = geneList, ont = "ALL", # "BP"、"MF"和"CC"或"ALL" OrgDb = OrgDb,#人类org.Hs.eg.db 鼠org.Mm.eg.db keyType = "ENTREZID", pvalueCutoff = 1) #实际为padj阈值可调整 GO_kk <- DOSE::setReadable(GO_kk_entrez, OrgDb=OrgDb, keyType='ENTREZID')#转化id save(KEGG_kk_entrez,KEGG_kk,GO_kk_entrez,GO_kk, file = "./Rdata/GSEA_result.RData") kk_gse <- KEGG_kk kk_gse_entrez <- KEGG_kk_entrez kk_gse_cut <- kk_gse[kk_gse$pvalue<1 & kk_gse$p.adjust<1 & abs(kk_gse$NES)>1] kk_gse_cut_down <- kk_gse_cut[kk_gse_cut$NES < 0,] kk_gse_cut_up <- kk_gse_cut[kk_gse_cut$NES > 0,] #选择展现NES前几个通路 down_gsea <- kk_gse_cut_down[tail(order(kk_gse_cut_down$NES,decreasing = T),10),] up_gsea <- kk_gse_cut_up[head(order(kk_gse_cut_up$NES,decreasing = T),10),] diff_gsea <- kk_gse_cut[head(order(abs(kk_gse_cut$NES),decreasing = T),10),] gseap1<-gseaNb(object = kk_gse, geneSetID = c(row.names(up_gsea)[1],row.names(down_gsea)[1]), subPlot = 3, addPval = T, pvalX = 1,pvalY = 2, curveCol=rainbow(10)#自动提供3种颜色,超过四个通路需要手动设置颜色 ) gseap1 width=15 height=7.5 ggsave(plot = gseap1,filename = './PDF/gseap1.pdf', width = width,height = height) ggsave(plot = gseap1,filename = './PNG/gseap1.png', width = width,height = height) gseap2 <- gseaNb(object = kk_gse, geneSetID = c(row.names(up_gsea)[c(1:5)],row.names(down_gsea)[c(1:5)]),#绘制哪几个通路的曲线 subPlot = 3,#保留几个副图 #termWidth = 35,#调整图例位置和长度 #legend.position = c(0.8,0.3),#调整图例位置和长度 addPval = F,#是否显示pval表格 pvalX = 0.05,pvalY = 0.05,#pval表格位置 curveCol=rainbow(10)#自动提供3种颜色,超过四个通路需要手动设置颜色 ) gseap2 width=15 height=7.5 ggsave(plot = gseap2,filename = './PDF/gseap2.pdf', width = width,height = height) ggsave(plot = gseap2,filename = './PNG/gseap2.png', width = width,height = height)
版权声明:本文内容由互联网用户自发贡献,该文观点仅代表作者本人。本站仅提供信息存储空间服务,不拥有所有权,不承担相关法律责任。如发现本站有涉嫌侵权/违法违规的内容,请联系我们,一经查实,本站将立刻删除。
如需转载请保留出处:https://51itzy.com/kjqy/38332.html