
关于单细胞的一些知识
Seurat 官方网站 https://satijalab.org/seurat/v3.2/pbmc3k_tutorial.html

讯享网
在官网教程中,作者选用的是PBMC的数据,可以从官网进行下载
下载后解压缩的文件夹包含三个文件
其中barcodes.csv是一个相当于每个细胞身份证的标记,在PBMC中总共有2700个单细胞,所以barcodes 总共有2700行,每一行代表一个细胞

genes.tsv呢,是一个ENSEMBL的ID 和一个Symbol 的ID ,总共有32738行
matrix.mtx是一个含有上述2700个细胞的Counts 数的矩阵,

上述的3个文件主要是从cellranger 流程下机后的数据可以通过Read10X这个函数进行读取,也可以将自己获得的单细胞矩阵的data.frame按照正常的文件读入的方式读进来,然后再创建Seurat 对象
library(dplyr) library(Seurat) library(patchwork) Load the PBMC dataset pbmc.data <- Read10X(data.dir ="../data/pbmc3k/filtered_gene_bc_matrices/hg19/")此处是自己解压后的PBMC的数据 # Initialize the Seurat object with the raw (non-normalized data). pbmc <- CreateSeuratObject(counts = pbmc.data, project = "pbmc3k", min.cells = 3, min.features = 200)通过CreateSeuratObject()创建一个Seurat对象,其包含矩阵数据和各类分析(如Data count 和PCA以及Cluster分析的结果 pbmc 讯享网
数据格式
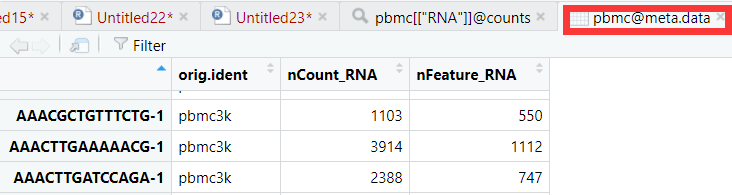
对于这个数据的标准分析流程
(一) 数据的前处理主要包括 数据的清洗,主要是去除低质量的细胞
依据QC质控进行过滤和选择细胞,数据归一化和缩放以及高变化基因特征的选择
一.QC和细胞的选择
1.常用的QC标准 是检测每个细胞中的Unique基因的数目,
a.低质量或者空的Droplet 通常含有很少的基因
b.双个或者多个细胞通常含有异常高的基因
2.每个细胞内总的分子数目也和Unique基因相似
3.线粒体基因组的占比
a.低质量或者死细胞里表现出异常多的线粒体基因组
b.通过PercentageFeatureSet()函数来计算每个特征基因集的占比
c.通过MT-开头来标注线粒体基因组
讯享网
QC的可视化,从而进行过滤细胞
过滤掉featire counts 超过2500或者小于200的细胞
过滤线粒体基因组占比大于5%的细胞
# Visualize QC metrics as a violin plot 通过小提琴图进行可视化,发现nFeature_RNA 的 VlnPlot(pbmc, features = c("nFeature_RNA", "nCount_RNA", "percent.mt"), ncol = 3) 
讯享网# FeatureScatter is typically used to visualize feature-feature relationships, but can be used # for anything calculated by the object, i.e. columns in object metadata, PC scores etc. plot1 <- FeatureScatter(pbmc, feature1 = "nCount_RNA", feature2 = "percent.mt") plot2 <- FeatureScatter(pbmc, feature1 = "nCount_RNA", feature2 = "nFeature_RNA") plot1 + plot2

数据的过滤 pbmc <- subset(pbmc, subset = nFeature_RNA > 200 & nFeature_RNA < 2500 & percent.mt < 5) (二) 数据的归一化
去掉了低质量的细胞之后,接下来就是数据的归一化,主要采用的是局部缩放的LogNormalize 的方法对每个细胞的总表达量乘以缩放因子(默认10000)对每个基因进行归一化。
讯享网采取的方法 pbmc <- NormalizeData(pbmc, normalization.method = "LogNormalize", scale.factor = 10000) 等同于这一步 pbmc <- NormalizeData(pbmc)
(三) 接下来去寻找高度可变的基因
主要是计算存在高变异的基因子集(在某些细胞中,这些基因高表达,在某些细胞中这些基因低表达)
此分析能够帮助下游进一步的聚焦生物学功能在单细胞集中。在Seurat3中作者采用的是FindVariableFeatures()函数寻找高度变异的基因。默认的情况下,作者在每个基因集里返回的是2000个基因的基因集。这将用于下游的PCA等分析。
pbmc <- FindVariableFeatures(pbmc, selection.method = "vst", nfeatures = 2000) # Identify the 10 most highly variable genes top10 <- head(VariableFeatures(pbmc), 10) # plot variable features with and without labels plot1 <- VariableFeaturePlot(pbmc) plot2 <- LabelPoints(plot = plot1, points = top10, repel = TRUE) plot1 + plot2 通过这一步的步骤,找到了高变异的 基因,并进行了注释

(四)接下来进行数据缩放
Seurat 的开发者采取的是线性转换,scaling ,这个方法是标准的处理步骤对于后续的降维操作,主要用的函数是ScaleData()
要点:
1.移动每个基因的表达,以使整个细胞的平均表达为0
2.对每个基因的表达进行缩放,使得每个细胞的变异系数是1,从而为下游的分析给出相同的权重,使得高表达的基因不占主导作用
3.这个结果主要在pbmc[[“RNA”]]@scale.data里面可以找到
讯享网Scaling the data all.genes <- rownames(pbmc) pbmc <- ScaleData(pbmc, features = all.genes) View(all.genes) View(pbmc[["RNA"]]@scale.data)
(五) 接下来进行线性降维
对缩放后的数据进行PCA降维处理,默认情况下,只是对先前的决定性的变化基因作为输入数据,但是如果想要改变这一默认,可以选择feature 这一参数,选择不同的基因集
pbmc <- RunPCA(pbmc, features = VariableFeatures(object = pbmc))进行PCA操作 对PCA进行可视化操作的方法 print(pbmc[["pca"]], dims = 1:5, nfeatures = 5) VizDimLoadings(pbmc, dims = 1:2, reduction = "pca")分别查看对PC1和PC2其主要作用的基因 DimPlot(pbmc, reduction = "pca")通过点图展示PC1和PC2 DimHeatmap(pbmc, dims = 1, cells = 500, balanced = TRUE)通过热图可以对PC成分进行可视化,允许我们对基因集的异质性进行审阅,从而决定哪个PC可用于后续的下游分析,一般默认可以选择500个细胞,细胞和基因都是按照PCA的分值进行排序的。 dims=1:15 画出1:15 的PC热图 

dimPlot(pbmc)后的图


(六)确定数据集的维度
讯享网pbmc <- JackStraw(pbmc, num.replicate = 100) pbmc <- ScoreJackStraw(pbmc, dims = 1:20) JackStrawPlot(pbmc, dims = 1:15)对结果进行可视化
1-12的PC 的Pvalue 都非常的小,小于0.005,因此这12个PC都是相对合理能够解释细胞变异的主成分,进一步的使用Elbowplot进行绘图,发现前7个成分能够解释大部分的变异,后面7以后的PC对标准离散值的都不大


(七)对于细胞的聚类
Seurat V3 采用的是基于图形的聚类方法,基于上一步的PC分析的距离矩阵用于后续的聚类分析。作者的这种基于PC分析,然后通过KNN的方法通过相似faeture的表达模式进行edges drawn,然后尝试将此图划分为高度相互联系的“半透明”或“社区”。
在 PhenoGraph里作者首次通过在PCA空间里基于欧氏距离构建KNN,并且重新定义了2个细胞的edge 权重(而这个是根据Jaccard similiarity),这一步主要是通过FindNeighbors()这个函数进行,输入先前确认好的维度的数据集(10个PC)
为了进一步的聚类细胞,接下来作者通过应用模块优化的方法(比如Louvain算法和SLM算法,将细胞迭代的组合在一起,以优化标准模块功能。 FindClusters()函数内置了这一过程,并包含了一个分辨率参数,通过改变granularity的值,可以改变下游的Cluster 的数目。参数越大,Cluster 的数目越多。
作者发现,参数在0.4-1.2时,通常可以约3K个单元,并且返回良好的结果。对于较大的数据集,**分辨率可能会增加,通过idens()函数可以找到Cluster.
pbmc <- FindNeighbors(pbmc, dims = 1:10) pbmc <- FindClusters(pbmc, resolution = 0.5) # Look at cluster IDs of the first 5 cells head(Idents(pbmc), 5) (八)非线性的降维(UMAP/tSNE)
Seurat 提供几种非线性的降维方法,主要是t-SNE 和UMAP,来可视化和探索数据集。这些算法的目标是学习数据的基础流形,以便将相似的细胞放在低维空间,基于图形聚类的细胞应该在降维图中共定位,作为t-SNE 和UMAP 的值输入,作者建议输入t-SNE 和UMAP 的数据应该和Cluster 分析的PCs 相同。
讯享网使用t-SNE 和UMAP进行可视化降维的结果 pbmc <- RunUMAP(pbmc, dims = 1:10)进行UMAP进行降维 pbmc <- RunTSNE(pbmc, dims = 1:10)进行UMAP进行降维 # note that you can set `label = TRUE` or use the LabelClusters function to help label # individual clusters DimPlot(pbmc, reduction = "umap",label = TRUE)

(九)寻找差异表达的Feature
Seurat 可以帮助你找到通过差异表达的基因作为marker 来定义的Cluster。通常默认的情况下,他可以帮助找到每一个Cluster 的阴参和阳参(通过ident.1),然后再于其他的细胞进行比较。FindAllMarkers()函数就可以帮助所有的Cluster 进行这一过程。min.pct参数需要一个feature 进行检测最小的百分比在2组细胞中。thresh.test 参数需要一个在2组差异表达的feature.可以设置这2个参数都为0,但是随着时间叫它将会急剧的增加,因为他将检测难以被高度区分的大量的feature,另一个选择是采用加速的算法max.cells.per.ident进行测试。这将采取向下取样的方法确定每一类的细胞。尽管这样将会失去它的作用,但是能够明显的增加速度,高度差异的基因将会被提升到Top 的位置。
# find all markers of cluster 1 cluster1.markers <- FindMarkers(pbmc, only.pos=TRUE,ident.1 = 1, min.pct = 0.25)查看第一个Cluster 的marker 基因min.pct是检测到的最小的百分比,nly.pos说明选取的是positive 的高变基因 head(cluster1.markers, n = 5) dim(cluster1.markers) View(cluster1.markers) # find all markers distinguishing cluster 5 from clusters 0 and 3 cluster5.markers <- FindMarkers(pbmc, ident.1 = 5, ident.2 = c(0, 3), min.pct = 0.25) head(cluster5.markers, n = 5) View(cluster5.markers) # find markers for every cluster compared to all remaining cells, report only the positive ones pbmc.markers <- FindAllMarkers(pbmc, only.pos = TRUE, min.pct = 0.25, logfc.threshold = 0.25) pbmc.markers %>% group_by(cluster) %>% top_n(n = 2, wt = avg_logFC) (十) marker 基因的可视化
Seurat 包对差异的基因的可视化,主要采取的是小提琴图VinPlot()。展示不同簇表达的分布,FeaturePlot() tSNE或者PCA作图基因表达的可视化,使用RidgePlot(),CellScatter 和DotPlot作为额外的方法来展现数据集。
讯享网VlnPlot(pbmc, features = c("MS4A1", "CD79A")) # you can plot raw counts as well VlnPlot(pbmc, features = c("FTL", "TFRC"), slot = "counts", log = TRUE)

FeaturePlot进行绘图
FeaturePlot(pbmc, features = c("MS4A1", "GNLY", "CD3E", "CD14", "FCER1A", "FCGR3A", "LYZ", "PPBP", "CD8A")) 
通过Doheatmap 进行可视化每一个簇已知基因的表达情况,通常每个簇选取20个marker进行展示
讯享网 top10 <- pbmc.markers %>% group_by(cluster) %>% top_n(n = 10, wt = avg_logFC) DoHeatmap(pbmc, features = top10$gene) + NoLegend()

(十一)每个簇的细胞类型的注释
在本包里,作者通过经典的marker基因进行了不同类的细胞的注释

通过对已知 的marker对细胞进行注释 new.cluster.ids <- c("Naive CD4 T", "Memory CD4 T", "CD14+ Mono", "B", "CD8 T", "FCGR3A+ Mono", "NK", "DC", "Platelet")向量注释基因名 names(new.cluster.ids) <- levels(pbmc)将基因名与Cluster 的因子进行一一对应 pbmc <- RenameIdents(pbmc, new.cluster.ids)利用RenameIdents进行重命名 DimPlot(pbmc, reduction = "umap", label = TRUE, pt.size = 0.5) + NoLegend()画图 
官网完整代码
讯享网library(SingleR) library(dplyr) library(Seurat) library(patchwork) BiocManager::install("SingleR") pbmc.data <- Read10X(data.dir = "/home/data/vip38/Data_resource/Single_Cell_Data_Test/hg19/") View(pbmc.data) pbmc <- CreateSeuratObject(counts = pbmc.data, project = "pbmc3k", min.cells = 3, min.features = 200) View() View(pbmc[["RNA"]]@counts) pbmc[["percent.mt"]] <- PercentageFeatureSet(pbmc, pattern = "^MT-")将每个细胞的线粒体基因占比进行分析 VlnPlot(pbmc, features = c("nFeature_RNA", "nCount_RNA", "percent.mt"), ncol = 3)通过小提琴图进行探索,样本整体的每个细胞中检测到的基因数目分布和RNA的Counts 分布以及下立体基因组的占比 # FeatureScatter is typically used to visualize feature-feature relationships, but can be used # for anything calculated by the object, i.e. columns in object metadata, PC scores etc. plot1 <- FeatureScatter(pbmc, feature1 = "nCount_RNA", feature2 = "percent.mt") plot2 <- FeatureScatter(pbmc, feature1 = "nCount_RNA", feature2 = "nFeature_RNA") plot1 + plot2将2个图拼在一起 pbmc <- subset(pbmc, subset = nFeature_RNA > 200 & nFeature_RNA < 2500 & percent.mt < 5)subset函数的使用,过滤掉Feature大于2500和小于200的 Normalization pbmc <- NormalizeData(pbmc, normalization.method = "LogNormalize", scale.factor = 10000) head() head(pbmc$) pbmc <- NormalizeData(pbmc)等同于上面一步 进行寻找高度可变的基因 pbmc <- FindVariableFeatures(pbmc, selection.method = "vst", nfeatures = 2000)方法是通过采用寻找平均变异系数进行分析 # Identify the 10 most highly variable genes top10 <- head(VariableFeatures(pbmc), 10)top10的高变异基因 head(top10)查看前10个的高变异基因 # plot variable features with and without labels plot1 <- VariableFeaturePlot(pbmc)画出标准变异系数和平均表达值之间的图 plot2 <- LabelPoints(plot = plot1, points = top10, repel = TRUE)对top10的基进行型标记 plot1 + plot2 Scaling the data all.genes <- rownames(pbmc) pbmc <- ScaleData(pbmc, features = all.genes) View(all.genes) View(pbmc[["RNA"]]@scale.data) Perform linear dimensional reduction pbmc <- RunPCA(pbmc, features = VariableFeatures(object = pbmc)) print(pbmc[["pca"]], dims = 1:5, nfeatures = 5) VizDimLoadings(pbmc, dims = 1:2, reduction = "pca")分别查看对PC1和PC2其主要作用的基因 DimPlot(pbmc, reduction = "pca") DimHeatmap(pbmc, dims = 1:2, cells = 500, balanced = TRUE)看前两个热图的结果 Determine the 'dimensionality' of the dataset pbmc <- JackStraw(pbmc, num.replicate = 100) pbmc <- ScoreJackStraw(pbmc, dims = 1:20) JackStrawPlot(pbmc, dims = 1:15)数据可视化 ElbowPlot(pbmc) 寻找cluster pbmc <- FindNeighbors(pbmc, dims = 1:10) pbmc <- FindClusters(pbmc, resolution = 0.5) head(Idents(pbmc), 5)前5个细胞的ID 使用t-SNE 和UMAP进行可视化降维的结果 pbmc <- RunUMAP(pbmc, dims = 1:10)进行UMAP进行降维 # note that you can set `label = TRUE` or use the LabelClusters function to help label # individual clusters DimPlot(pbmc, reduction = "umap",label = TRUE) saveRDS(pbmc, file = "pbmc_tutorial.rds") # find all markers of cluster 1 cluster1.markers <- FindMarkers(pbmc, ident.1 = 1, min.pct = 0.25)min.pct是检测到的最小的百分比 head(cluster1.markers, n = 5) dim(cluster1.markers) View(cluster1.markers) # find all markers distinguishing cluster 5 from clusters 0 and 3 cluster5.markers <- FindMarkers(pbmc, ident.1 = 5, ident.2 = c(0, 3), min.pct = 0.25) head(cluster5.markers, n = 5) dim(cluster5.markers) View(cluster5.markers) # find markers for every cluster compared to all remaining cells, report only the positive ones pbmc.markers <- FindAllMarkers(pbmc, only.pos = TRUE, min.pct = 0.25, logfc.threshold = 0.25) pbmc.markers %>% group_by(cluster) %>% top_n(n = 2, wt = avg_logFC) cluster1.markers <- FindMarkers(pbmc, ident.1 = 0, logfc.threshold = 0.25, test.use = "roc", only.pos = TRUE) VlnPlot(pbmc, features = c("FTL", "HEPH")) # you can plot raw counts as well VlnPlot(pbmc, features = c("FTL", "TFRC"), slot = "counts", log = TRUE) FeaturePlot(pbmc, features = c("MS4A1", "GNLY", "CD3E", "CD14", "FCER1A", "FCGR3A", "LYZ", "PPBP", "CD8A")) top10 <- pbmc.markers %>% group_by(cluster) %>% top_n(n = 10, wt = avg_logFC) DoHeatmap(pbmc, features = top10$gene) + NoLegend() 已知基因的注释 new.cluster.ids <- c("Naive CD4 T", "Memory CD4 T", "CD14+ Mono", "B", "CD8 T", "FCGR3A+ Mono", "NK", "DC", "Platelet") names(new.cluster.ids) <- levels(pbmc) pbmc <- RenameIdents(pbmc, new.cluster.ids) DimPlot(pbmc, reduction = "umap", label = TRUE, pt.size = 0.5) + NoLegend() saveRDS(pbmc, file = "pbmc3k_final.rds")
版权声明:本文内容由互联网用户自发贡献,该文观点仅代表作者本人。本站仅提供信息存储空间服务,不拥有所有权,不承担相关法律责任。如发现本站有涉嫌侵权/违法违规的内容,请联系我们,一经查实,本站将立刻删除。
如需转载请保留出处:https://51itzy.com/kjqy/63364.html