
点击蓝字 关注我们
MetOrigin:代谢物溯源推动肠道微生物和代谢整合分析

https://doi.org/10.1002/imt2.10

2022/3/21
● 2022年3月21日,浙江大学医学院附属儿童医院国家儿童健康与疾病临床研究中心倪艳团队在iMeta在线发表题为“MetOrigin: Discriminating the origins of microbial metabolites for integrative analysis of the gut microbiome and metabolome”的研究型文章。
● 该文章开发了一个交互式分析软件MetOrigin,能够对代谢产物进行溯源分析。不仅可以快速识别微生物来源代谢物及其代谢功能,还有助于发现与其密切相关的关键微生物。
● 第一作者:俞刚、徐翠芳
● 通讯作者:倪艳
()
摘 要
肠道微生物和代谢的交互作用在人类健康和疾病中起着重要作用。当前的研究主要是利用统计分析方法来挖掘肠道微生物和代谢的相关性。但是,在没有体外培养实验进行验证的情况下,很难确定微生物的代谢功能。我们认为,通过鉴定代谢物的来源(如宿主、食物或环境),分析不同来源代谢物的代谢功能及其与微生物的关联性,能够提高发现生物标记物的效率和准确性。目前尚缺少相关的生物信息学分析工具。因此,我们开发了一个交互式分析软件MetOrigin,能够对代谢产物进行溯源分析,分别进行代谢通路富集分析,并通过Sankey网络图整合和展示微生物和代谢物在统计学和生物学层面有意义的关联性。MetOrigin不仅能够快速识别微生物来源代谢物及其代谢功能,还有助于发现与其密切相关的关键微生物。
MetOrigin 的访问链接是:
http://metorigin.met-bioinformatics.cn/
关键词:肠道微生物和代谢物、相关性分析、代谢物来源、Sankey网络、代谢通路富集分析
亮 点
● MetOrigin能够快速鉴定微生物相关代谢产物和分析其代谢功能
● MetOrigin使用Sankey网络整合统计学和生物学关联性,鉴定关键代谢产物的相关微生物菌种
● MetOrigin是一款微生物组和代谢物组整合分析的在线软件
(公开链接方式:http://metorigin.met-bioinformatics.cn/)
视频解读
Youtube:https://youtu.be/lz4WbG9VD5w
中文翻译、PPT、中/英文视频解读等扩展资料下载
请访问期刊官网:http://www.imeta.science/
全文解读
引 言
代谢组和微生物组已应用于很多人类疾病研究,包括肥胖、炎症性肠病、心血管疾病、神经退行性疾病和癌症等[1]。肠道微生物通过饮食、药物和宿主代谢物等(如短链脂肪酸、氨基酸和胆汁酸等)产生各种不同的小分子代谢物,并进一步参与和维持宿主生理活动[2]。另一方面,这些代谢物能够维持微生态平衡,参与宿主能量代谢和调控免疫功能等[3, 4]。很多研究报道微生物来源的代谢物可以作为疾病诊断和预防的生物标记物[2]。尽管宿主和微生物环境中的代谢产物来源复杂,我们大致可以概括3种代谢物的来源:微生物从头合成,饮食来源经微生物代谢,宿主来源经微生物修饰[5]。然而,目前的研究和知识还不能完全理解微生物的代谢能力,微生物如何参与代谢反应过程,甚至很难区分代谢产物是否来源于微生物还是宿主。
很多研究通过同时开展代谢组学和宏基因组学分析,来探索代谢产物和微生物之间的相关性[6]。然后,通过大量的文献检索和体内外实验对生物学功能进行验证。因此,该研究领域迫切需要生物信息学分析方法和工具,整合有价值的先验生物学知识和统计分析结果,来挖掘微生物和代谢物之间的相互作用。目前已发表的一些软件,例如MIMOSA可以预测生态群落对代谢物浓度的影响[7],AMON可以区分微生物组或宿主对代谢物的影响[8]。这些研究初步探索并强调不同物种对代谢的贡献不同。但是,没有进一步区分属于宿主或微生物群落的代谢功能。如果把来自宿主和微生物群的代谢物混在一起,我们很难鉴别出微生物代谢物的真正生物学功能。此外,微生物与代谢物之间的统计学相关性分析可以指导我们探索新机制,如果结合生物学意义就能更好地帮助我们了解生物体的代谢活动。因此,我们提出将统计学和生物学关联结合起来,探索复杂的微生物和代谢物互作网络。
本研究介绍了一个生物信息学分析流程—MetOrigin,通过搜索数据库来对代谢物溯源,根据不同来源进行代谢通路富集分析(MPEA),利用强大的Sankey网络将生物学和统计学层面的相关性整合和可视化,最终帮助研究人员提出微生物-代谢物相互作用的科学假设。我们利用两项肥胖相关肠道微生物组和代谢物组学研究验证MetOrigin的可行性,包含了一项肥胖儿童队列和TwinsUK队列研究。MetOrigin可以通过以下网址进行访问:http://metorigin.met-bioinformatics.cn/
方 法
两项肥胖患者的肠道微生物组学和代谢物组学研究
● 研究一:肥胖儿童队列
研究共纳入11名肥胖儿童(BMI指数z值≥ 2)和 17名体重正常儿童。使用Illumina Novaseq 6000平台对粪便样本进行宏基因组测序。依照文献报道中的方法,对血清样品进行初步处理,以提取代谢物并去除蛋白质[9],然后使用超高效液相色谱/串联质谱(UPLC-MS/MS)平台对上清液进行非靶向代谢物组学分析。
● 研究二:TwinsUK队列
我们分析了来自TwinsUK队列中786名个体的粪便代谢物组和微生物组[10]。依照文献报道中的方法,从粪便样本中提取16S rRNA,进行PCR扩增,对每个样本进行条形码编码,并使用Illumina MiSeq平台进行测序[11]。使用UPLC-MS/MS平台对粪便样本进行非靶向代谢物组学分析[12]。
代谢物来源数据库
MetOrigin整合了7个代谢物数据库,这些数据库含有代谢物来源的信息,包括京都基因和基因组百科全书(KEGG)[13]、人类代谢物组数据库(HMDB)[14]、BIGG[15]和ChEBI[16]、食品数据库(FoodDB)[17]、药物数据库[18],以及毒素和毒素靶标数据库(T3DB)[19](图1A)。首先,我们合并了7个数据库中的所有代谢物。KEGG、HMDB、BIGG和ChEBI数据库包含了来自哺乳动物、微生物、食品、药物、毒素和污染物等的代谢物,而FoodDB、Drugbank和T3DB相应地包含了来自食品、药物和毒素的代谢物。然后,根据数据库ID、化学式和化合物名称或同义词,对来自不同数据库的代谢物进行交叉验证,进一步检查和去除冗余代谢物。目前,MetOrigin数据库有个“非冗余”代谢物,每种代谢物至少拥有一个数据库的链接。其中种代谢物含有明确的来源信息,可分为6类,包括宿主(哺乳动物)、微生物(古生菌、真菌、细菌)、宿主-微生物共代谢(宿主和微生物群共享)、食物(食物和植物)、药物和环境(毒素和污染物)。表S1中列出了7个数据库的详细信息(包括更新日期和代谢物来源)。使用MetOrigin,代谢物组学研究中的特定代谢物可以根据其KEGG ID、HMDB ID和/或化合物名称(包括所有同义词)进行匹配,然后分类为不同来源,为后续分析做准备(图1B)。
基于代谢物来源的代谢功能分析
MPEA代谢通路富集分析已常用于代谢物组学研究中[20]。通常,该方法是对所有的差异代谢物进行处理。但是,MetOrigin是分别对来自不同来源的差异代谢物进行MPEA分析。而且,MEPA分析所需的代谢通路参考数据库也分别选择来自宿主、微生物群或两者共有的三种类型。宿主的参考代谢通路是来自KEGG数据库(如人类和小鼠等)。微生物群落的参考代谢通路是来自数据库中6800多个微生物。宿主和微生物共有的参考代谢通路则是两者的整合。
利用Sankey网络分析整合生物学与统计学相关性
MetOrigin用两种不同的方式挖掘微生物和代谢物组的相关性:统计学和生物学意义上的关联分析。首先,MetOrigin提供了3种经典的相关性分析方法进行统计学分析,包括Spearman、Pearson和最大信息系数(MIC)分析。其次,我们利用KEGG数据库搜索可能参与相关代谢反应的微生物,并与代谢物进行关联。最后,我们利用Sankey网络分析,直观地展示这两层相关性[21, 22]。Sankey网络图主要有两种展示方式。在Bio-Sankey网络中,我们探索了所有可能参与某个代谢反应的微生物,并在不同分类水平(即门、纲、目、科、属和种)将细菌与代谢物进行关联。Bio-Sankey网络旨在探索细菌与代谢物(及其代谢反应)之间的生物学相关性,同时能得到统计学验证。在STA-Sankey网络中,我们整合了所有与代谢反应有统计学关联的细菌,如果存在生物学关系,它们会以红色或绿色突出显示。与Bio-Sankey网络相比,STA-Sankey关注真实数据集中细菌和代谢物之间的统计学相关性,解释它们之间的潜在关系。
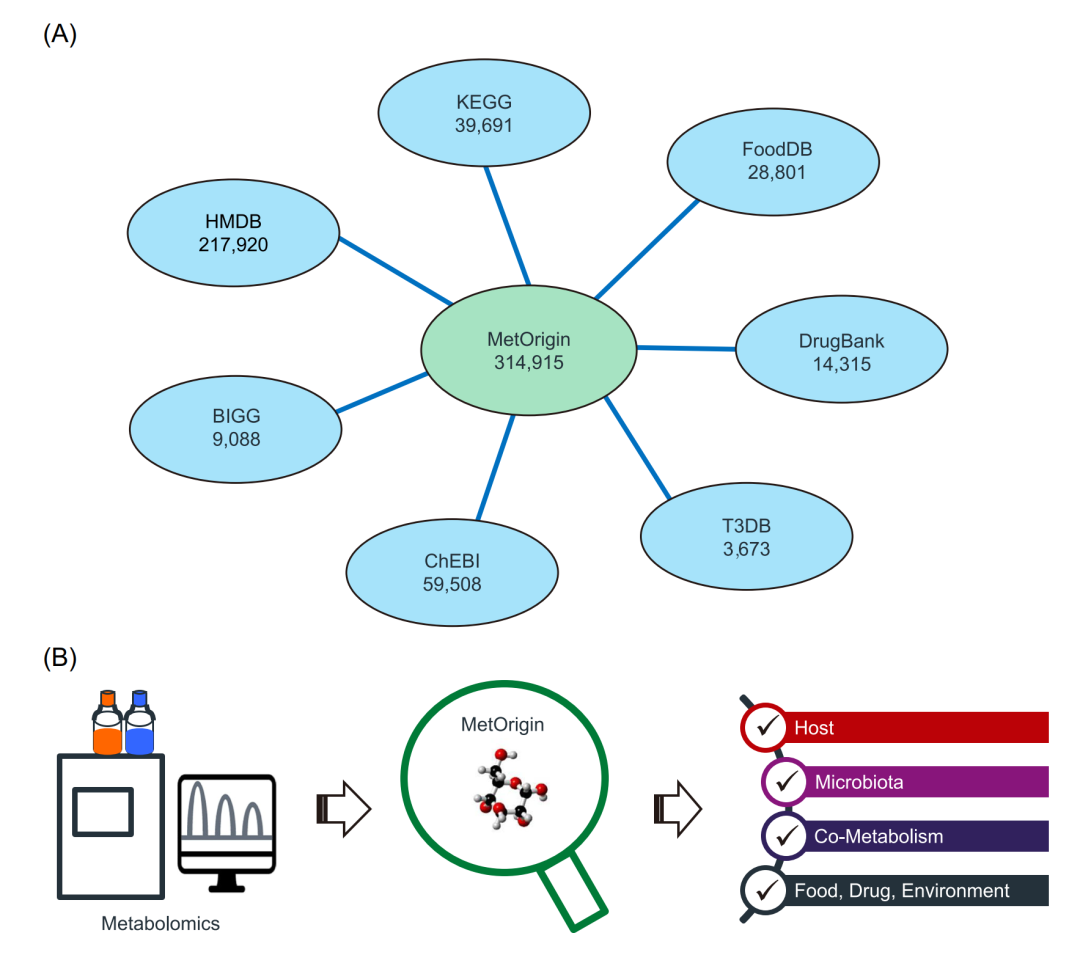
图1 MetOrigin代谢物数据库和溯源分析
(A)MetOrigin中7个代谢物数据库。(B) MetOrigin代谢物溯源分析。
结 果
MetOrigin工作流程
MetOrigin是一个可视化交互式的应用软件,对微生物和代谢物组数据进行整合分析(图S1)。MetOrigin根据研究目的,提供了了两种类型的数据分析模式。第一种是简单MetOrigin分析(Simple MetOrigin Analysis,简称SMOA),需要提供包含KEGG或HMDB ID的代谢物列表。SMOA模式提供了代谢物溯源分析,代谢通路富集分析,以及Sankey网络图分析来探索微生物和代谢物的生物学相关性。第二种模式是深度MetOrigin分析(Deep MetOrigin Analysis,简称DMOA),需要3种类型的数据,包括(1)代谢物列表,其中包含化合物名称、KEGG或HMDB ID以及丰度/浓度值;(2)微生物组列表,包括微生物注释和来自16S rRNA基因测序或宏基因组测序的丰度值;(3)样本名称和分组信息的样本信息表,例如包含对照组与患病组的信息。除了代谢物溯源分析、代谢功能分析和Sankey图外,DMOA还提供相关性分析和网络总结分析,来进一步探索微生物和代谢物在统计学和生物学意义上的相关性。
在进行统计分析之前,需要对数据进行预处理,这是代谢组学和微生物组学研究中的一个重要步骤。数据预处理包括缺失值插补和数据标准化。在我们之前的研究中比较和评估了各种缺失值处理方法的特点,包括最小值替换缺失值、应用随机森林(RF)、K近邻(KNN)或左删失数据分位数回归插补法(QRILC)[23]。我们在MetOrigin中提供了上述方法,用户需仔细选择一种合适方法,并评估其对统计分析的影响。同时,MetOrigin使用了数据转换和百分比计算方法对数据进行归一化处理。在MetOrigin中完成所有分析后,所有数据分析结果可以下载,包括表格和高分辨率的图,用于进一步的数据分析和挖掘。
MetOrigin数据库
MetOrigin从7个代谢物数据库中收集了共个“非冗余”代谢物(图1A)。其中种代谢物包含了来源信息,包括宿主(哺乳动物)、微生物群(古生菌、真菌、细菌)、共代谢(宿主和微生物群共享)、食品(食品和植物)、药物、和环境(毒素和污染物)(图1B)。MetOrigin从KEGG数据库中获取7326种不同的物种,其中有6382种细菌微生物和285种动物(图2A)。我们计算了属于人类(hsa)的代谢物和富集代谢通路的总数,并将其与微生物群的代谢物与代谢通路进行了比较(图2B和C)。结果显示有426种人类特有的代谢产物,共参与8条代谢通路,前5条代谢通路是类固醇激素生物合成(steroid hormone biosynthesis,)、药物代谢(drug metabolism)、外源性物质代谢(metabolism of xenobiotics,)、鞘糖脂生物合成(glycosphingolipid biosynthesis)和初级胆汁酸生物合成(primary bile acid biosynthesis)(图2D)。另外,微生物特有的2099种代谢产物共参与64条代谢通路,前5条代谢通路是类胡萝卜素生物合成(carotenoid biosynthesis)、卟啉和叶绿素代谢(porphyrin and chlorophyll metabolism)、II型聚酮产物合成(biosynthesis of type II polyketide products)、氨基糖和核苷酸糖代谢(amino sugar, and nucleotide sugar metabolism),以及O-抗原核苷酸糖生物合成(O-antigen nucleotide sugar biosynthesis)(图2F)。同时,有1217种代谢产物是人类和微生物群落共有,共参与81种代谢通路,前5种富集的代谢通路是嘌呤代谢(purine metabolism)、氨基酰基tRNA生物合成(aminoacyl-tRNA biosynthesis)、脂肪酸生物合成(fatty acid biosynthesis,)、嘧啶代谢(pyrimidine metabolism)和脂肪酸降解(fatty acid degradation)(图2E)。
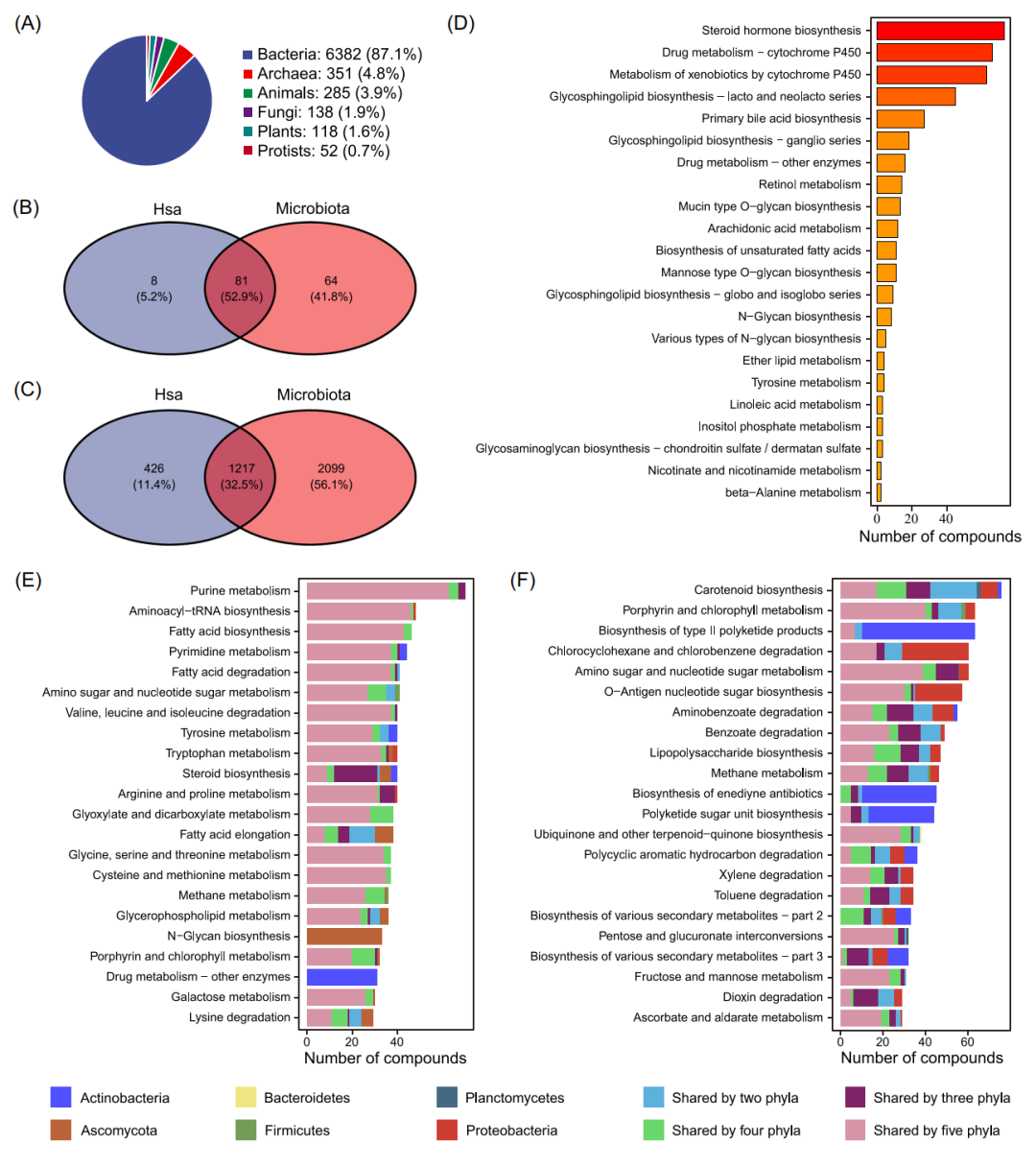
图2. MetOrigin数据库总结
(A) KEGG数据库中不同物种总结。(B) 人类和微生物群落中代谢通路韦恩图。(C) 人类和微生物群落中代谢物数量韦恩图。(D-F)人类、微生物和(E)两者共有的富集代谢通路的条形图。
肥胖儿童的肠道微生物和代谢物组整合分析
● 代谢物溯源分析和基于代谢物来源的代谢功能分析
通过MetOrigin分析,我们将923种代谢物分为4类:16种宿主(人类)来源代谢物、103种细菌来源代谢物、204种细菌-宿主共有的代谢物,以及600种属于其他来源(药物或食品等)(图3A)。通过差异分析得到249种代谢物与肥胖疾病相关,包括6种宿主来源代谢物、32种细菌来源代谢物、65种细菌-宿主共代谢的代谢物以及146种其他来源代谢物。进一步,我们将这些差异代谢物根据来源选择对应的参考代谢通路数据库进行MPEA分析。并且,我们也采用传统的方法进行分析和比较,直接使用人类代谢通路数据库做参考,或直接把人类和细菌代谢通路数据库整合做参考(图3C)。通过对比,我们发现,直接使用人类代谢通路数据库进行MPEA分析会忽略微生物代谢产物的贡献。然而,将人类和细菌相关通路整合作为参考又会带来重叠效应。相比之下,基于代谢物溯源的MPEA分析能根据代谢物来源的不同明确他们各自的代谢功能。在儿童肥胖研究中,我们分别得到2、14和74条与宿主、细菌和共代谢相关的代谢通路(图3B)。其中,宿主、细菌和共代谢中分别有2、2和31条代谢通路与肥胖显著相关(P<0.05)。烟酸和烟酰胺代谢(nicotinate and nicotinamide metabolism)以及类固醇激素生物合成是宿主特有。苯丙氨酸代谢(phenylalanine metabolism)和嘌呤代谢是微生物群落特有,而宿主和微生物群共有31条与氨基酸、脂质和糖类相关的代谢途径。
● 使用Sankey网络图展示微生物和代谢物之间生物学和统计学相关性
MetOrigin使用Spearman分析对肠道微生物和代谢物之间进行相关性分析。在门、纲、目、科、属和种水平上,分别有902、 1364、2431、4113、13285和62976对细菌和代谢产物密切相关(P<0.05)。根据前面的分析,我们确定了两条微生物特有的代谢通路与肥胖相关,即苯丙氨酸代谢和嘌呤代谢。以苯丙氨酸代谢通路为例,该代谢途径涉及4种不同的代谢物,即苯甲酸盐(benzoate)、苯乳酸(phenyllactate)、N-乙酰苯丙氨酸(N-acetylphenylalanine)和3-羟基苯丙酸盐(3-Hydroxyphenylpropanoate),共参与5种不同的代谢反应(R00693、R01370、R01371、R01424和R06786)。对于每个代谢反应,MetOrigin利用Bio-Sankey和STA-Sankey网络分析微生物和代谢物之间的生物学和统计学相关性。在Bio-Sankey网络中,与代谢反应R00693密切相关的是变形菌(Proteobacteria)和厚壁菌(Firmicute)(图4A)。苯丙氨酸(phenylalaine)通过乙酰辅酶A:L-苯丙氨酸N-乙酰转移酶(Acetyl-CoA: L-phenylalanine N-acetyltransferase)产生N-乙酰苯丙氨酸(N-Acetyl-phenylalanine)。在这项研究中,我们发现苯丙氨酸(phenylalaine)和N-乙酰苯丙氨酸(N-Acetyl-phenylalanine)在肥胖中显著上升。克雷伯氏菌(Klebsiella)、耶尔森菌(Yersinia)和梭菌(Clostridium)是肥胖患者中最显著增加的菌属(P<0.05,深红色),它们与R00693代谢反应密切相关。在STA-Sankey网络中,统计分析证实变形菌和厚壁菌与代谢反应R00693中的苯丙氨酸和N-乙酰苯丙氨酸密切相关,这与Bio-Sankey的结果是互补的(图4B)。总之,Bio-Sankey和STA-Sankey网络分析可以提供微生物和代谢物组之间的生物学和统计学的相关性。
● 网络总结分析
最后,我们分别整合来自宿主、微生物群和共代谢来源的差异代谢物,以及相关细菌,以获得与肥胖相关的微生物和代谢物相互作用网络。宿主特有的代谢网络表明,烟酸和烟酰胺代谢通路和类固醇激素生物合成通路中有5种代谢产物显著上调(图5A)。苯丙氨酸代谢网络表明,通过生物学和统计相关分析验证,共有7种代谢物与6种菌属密切相关(P<0.01)(图5B)。宿主-微生物共代谢网络显示,牛磺酸和低牛磺酸代谢(taurine and hypotaurine metabolism)有8种代谢产物与17种不同的细菌相关(P<0.01),其中大多数呈负相关(图5C)。总之,宿主特异性代谢网络和微生物-代谢物关联网络提供了与肥胖相关的更具体的代谢变化信息。
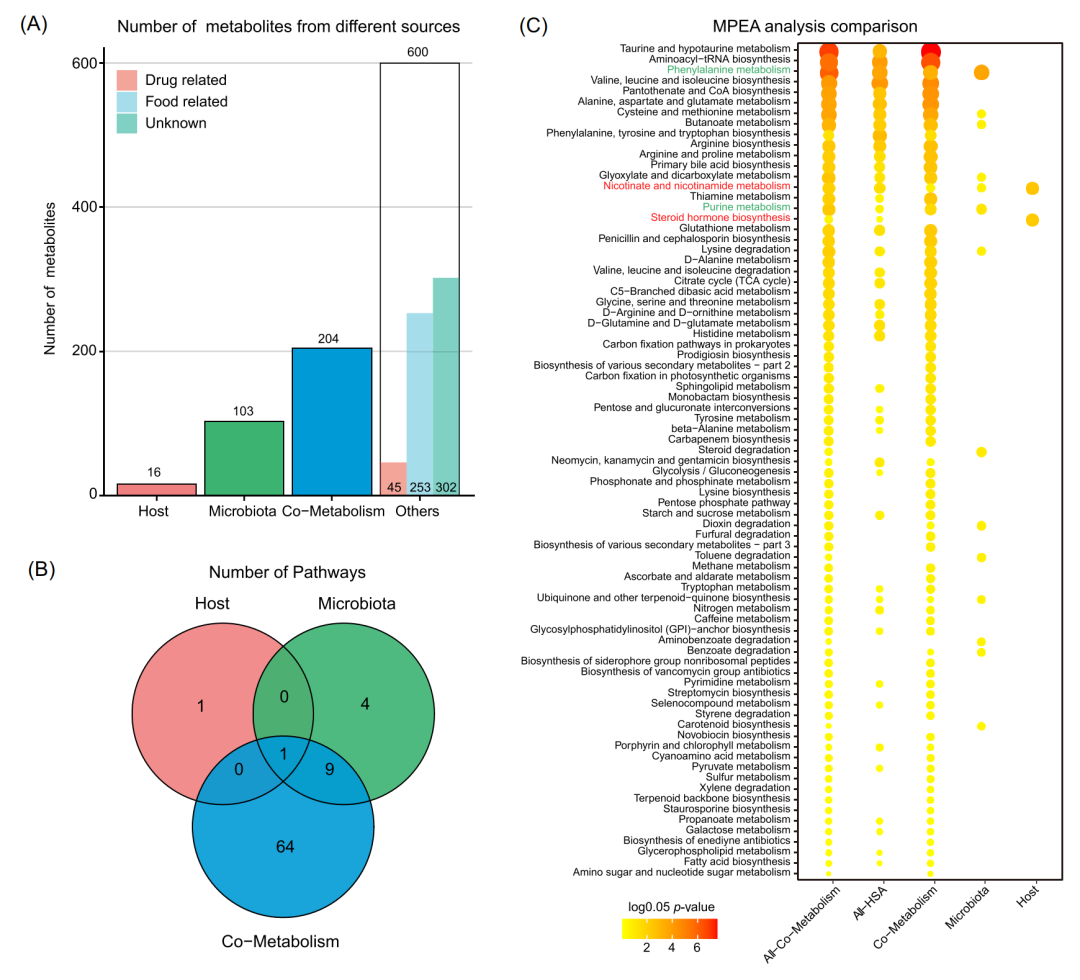
图3. 肥胖儿童队列的代谢物溯源分析和基于代谢物来源的功能分析
(A) 不同来源的代谢物数量条形图。(B) 基于代谢物来源的MPEA分析所得到的代谢通路韦恩图。(C) 使用不同方法进行MPEA分析和比较,包括应用来自宿主(Host)、微生物群(Microbiota) 或共代谢(Co-Metabolism)的代谢物亚组,并将它们匹配到相应的路径上,或以传统方式将所有代谢物匹配到宿主来源(ALL-HSA)或宿主-微生物共有的代谢通路(All-Co-Metabolism)。
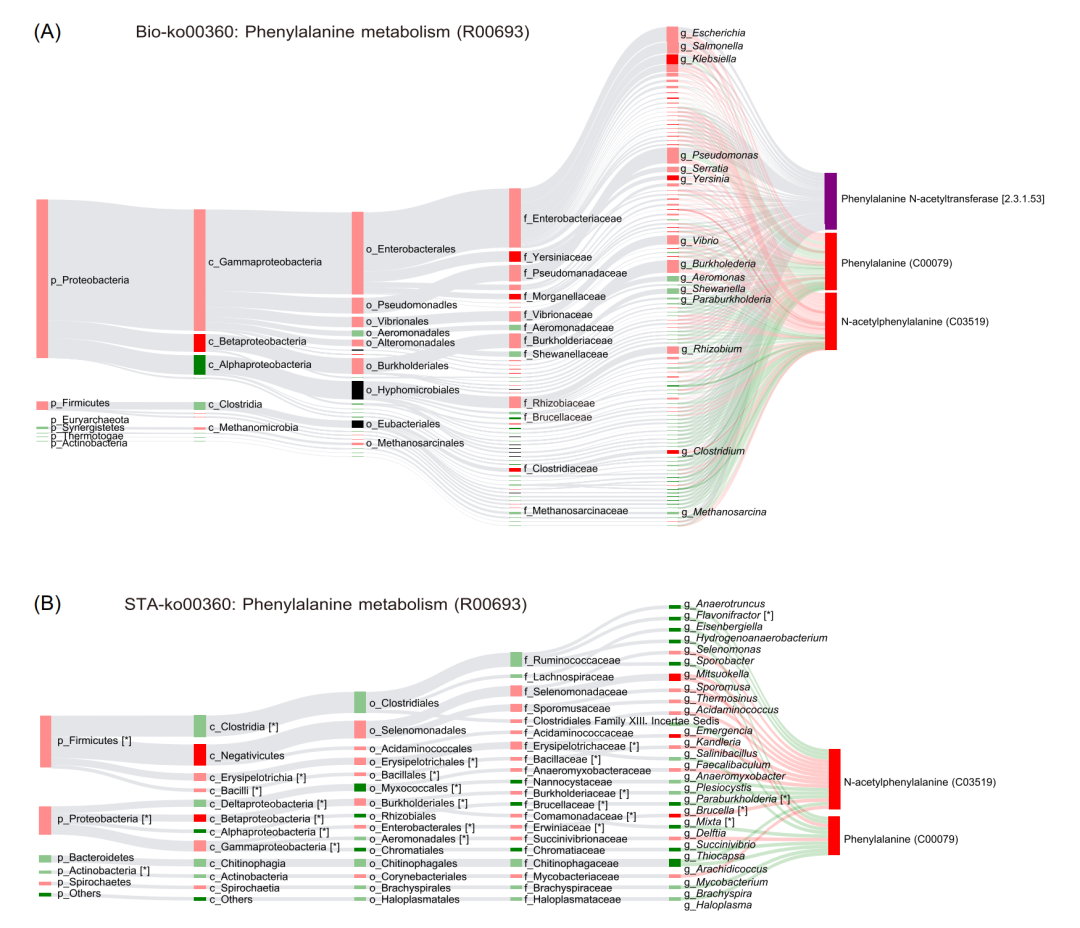
图4. 苯丙氨酸代谢途径网络图
(A)苯丙氨酸代谢途径(R00693)的BIO-Sankey网络图。(B) 苯丙氨酸代谢途径(R00693)的STA-Sankey网络。星号(*)表示与代谢物的有统计学意义的相关性。红色(或绿色)节点表示上调(或下调)。红色(或绿色)条带表示与代谢物的正(或负)相关性。深红色/绿色表示P<0.05。
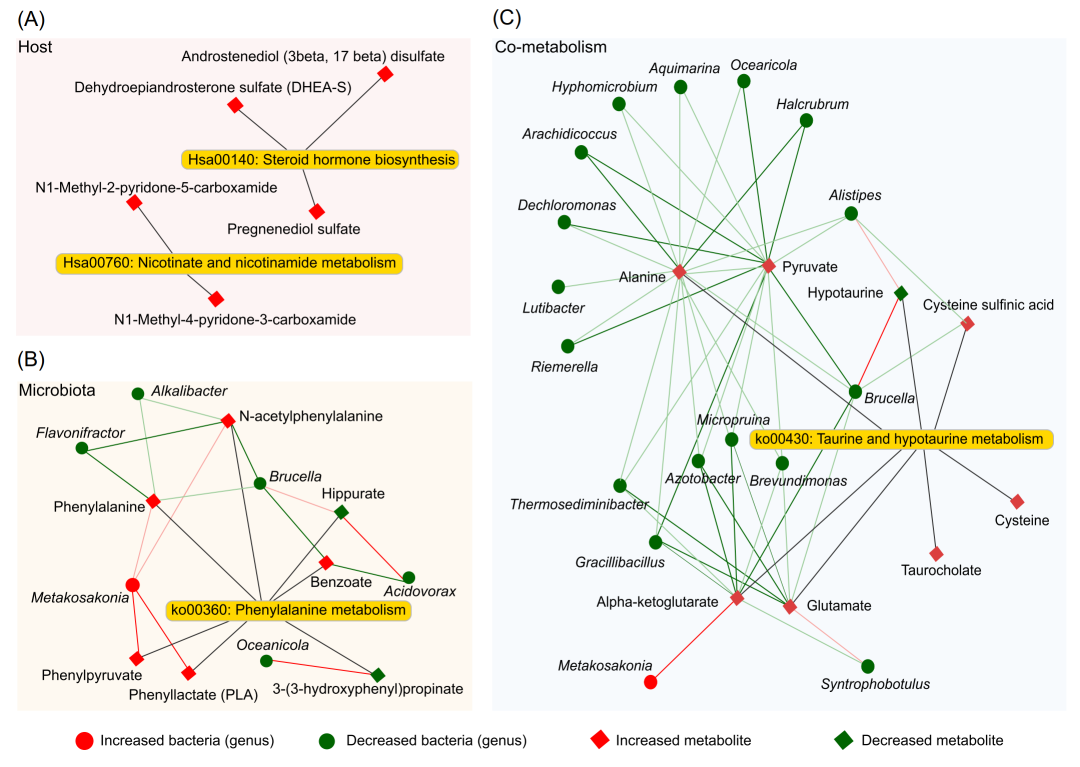
图5. 肥胖儿童队列的网络总结分析
(A)宿主来源网络;(B)微生物群来源网络;(C)宿主和微生物共有网络。菱形和圆点形状分别表示相关的代谢产物和微生物。红色(或绿色)节点表示上调(或下调)。红(或绿)的连接线表示微生物和代谢物之间的正(或负)相关性。
TwinsUK队列肠道微生物组和代谢物组整合分析
我们使用MetOrgin对TwinsUK队列中肥胖患者的肠道微生物组和代谢组进行分析。总共923种代谢物分为4组:11种宿主来源代谢物、127种细菌来源代谢物、224种细菌-宿主共代谢物和487种其他来源的代谢物(图6A)。其中,根据BMI指数将人群分为两组进行比较(BMI<24 vs BMI>28),差异分析得到93种差异代谢物与肥胖相关,包括3种宿主来源代谢物、19种细菌来源代谢物、23种细菌-宿主共代谢物和48种其他来源的代谢物。基于代谢来源的MPEA分析结果显示,宿主、微生物群以及共代谢的代谢途径分别2条、14条和42条(图6B)。与儿童肥胖研究类似,类固醇激素生物合成是宿主特有的。在42种宿主和细菌共有的代谢途径中,有3种与氨基酸相关代谢,分别为精氨酸生物合成(Arginine biosynthesis),牛磺酸和低牛磺酸代谢,丙氨酸、天冬氨酸和谷氨酸代谢(Alanine, aspartate and glutamate metabolism),这3类氨基酸代谢差异在儿童和成人肥胖研究中都具有统计学差异。微生物与代谢产物的相关性进一步通过网络图进行可视化(图6C和6D)。
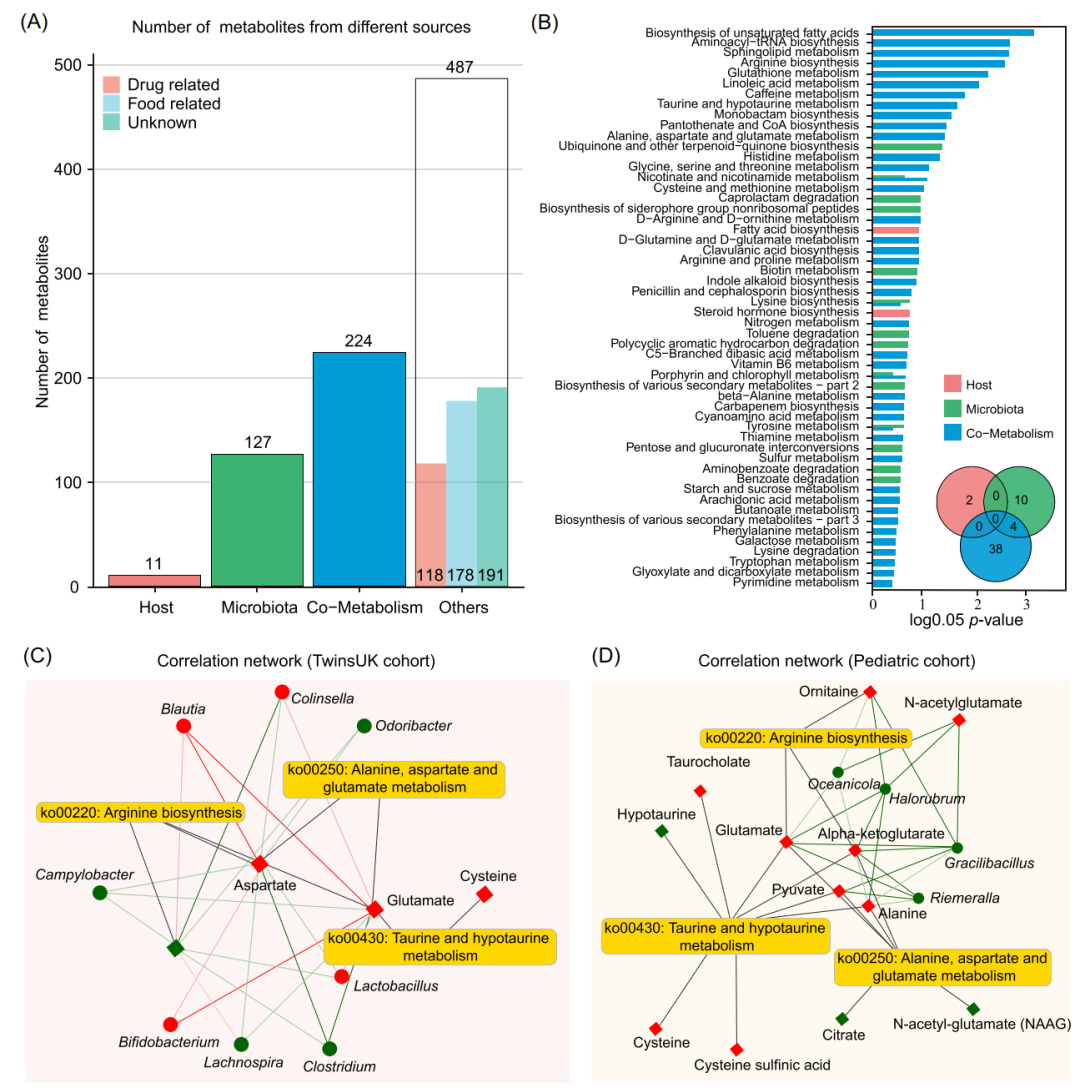
图6.TwinsUK队列研究的MetOrigin分析
(A) 不同来源代谢物数量的条形图。(B) 基于代谢物来源的MPEA分析得出的富集代谢通路条形图和维恩图。(C)TwinsUK队列研究中3种代谢通路中微生物和代谢物的相关性网络图。(D)儿童肥胖队列研究中3种代谢通路中微生物和代谢物的相关性网络图。
讨 论
近年来,人类健康和疾病的肠道微生物和代谢物组研究不断增加,然而微生物和代谢物之间的相互作用有待进一步探索和挖掘。目前的分析策略主要有:1.从代谢物组学研究中识别来自微生物的代谢产物(如短链脂肪酸、支链脂肪酸和胆汁酸),表明微生物可能参与疾病的相关代谢过程[2];2.用宏基因组测序结果预测微生物群落的代谢功能[24],并对代谢组和宏基因组进行统计学相关性分析[25, 26]。然而,对参与代谢反应的关键微生物进行验证是一个费时费力的过程。MetOrigin旨在通过生物信息分析技术,加速微生物-代谢物互作关系验证的过程。总的来说,MetOrigin通过代谢物溯源,根据代谢物来源进行功能分析,整合统计和生物学意义的相关性结果,最后得到具有较高准确性的生物标志物。
MetOrigin分析的第一步是鉴定代谢物的不同来源。目前,软件对代谢物来源的确定主要取决于7个数据库,包括HMDB、BIGG和ChEBI、FoodDB、Drugbank和T3DB。到目前为止,MetOrigin包含了共种含有特定来源信息的代谢物,这些来源包括宿主(哺乳动物)、微生物群(古生菌、真菌、细菌)、共代谢(宿主和微生物群共享),或来自食物(食物和植物)、药物和环境(毒素和污染物)。未来,我们将进一步探索和整合其他数据库和文献中的有用信息,提高软件对代谢物来源鉴别的准确性和可靠性,为进一步的科学研究和发现提供帮助。
Sankey分析是MetOrigin的核心,旨在阐明代谢物和相关微生物之间的生物学和统计学上的相关性。对于Bio-Sankey图,我们会列出在数据库中与已知代谢反应相关的所有细菌。在微生物不同分类水平, Sankey网络图的条带宽度反应细菌和代谢物之间关联性程度,并同时标注具有统计学差异的相关性。而根据STA-Sankey网络分析,我们可以提供高度相关的微生物和代谢物,挖掘高通量大数据中新的微生物和代谢关系。Bio-Sankey和STA-Sankey的结合可以帮助我们更快速的发现潜在的生物标记物,并为进一步的科学研究提供新的假设。
MetOrigin旨在有效探索肠道微生物和代谢组的交互作用,并准确识别微生物和代谢物之间的相关性。然而,关键细菌是否真正参与相关代谢反应,或者细菌是否利用或产生某种代谢物,仍然需要通过无菌动物研究或体外微生物培养模型来进一步验证。最后,MetOrigin不局限于对肠道微生物组的研究,也可以探索其他微生物组(如口腔、皮肤、环境等)与代谢物之间的相关性。
结 论
MetOrigin是一个公开、可视化交互式的应用软件,。它可以帮助没有生物信息学背景的科学家简化数据挖掘的过程。MetOrigin满足了研究者对探索微生物组和代谢物组之间复杂关系的迫切需要。MetOrigin提出了一种新颖的分析方法:将来自不同来源的代谢物进行代谢途径富集分析,并使用Sankey网络将具有生物学和统计学意义的相关微生物与代谢物进行关联。这为科研工作者提供了与微生物相关的代谢信息,并帮助他们从微生物和代谢平衡的角度探索新的饮食或药理学干预策略。
致谢:感谢国家重点研发计划(2021YFC27 0804)和国家自然科学基金(、)的资助。感谢TwinsUK研究组分享的宝贵数据。
引文格式:Yu, Gang, Cuifang Xu,Danni Zhang, Feng Ju, and Yan Ni. 2022.“MetOrigin: Discriminating the Origins of Microbial Metabolites for Integrative Analysis ofthe Gut Microbiome and Metabolome.”iMeta. e10. https://doi.org/10.1002/imt2.10

作者简介

倪艳(通讯作者)
● 浙江大学医学院附属儿童医院,特聘研究员,博士生导师。
● 2013-2017年担任美国夏威夷大学癌症研究中心助理教授,博士生导师,在癌症流行病学和代谢组学中心工作。分别于2006年获得华东理工大学制药工程学士学位、2009年获得上海交通大学药剂学医学硕士学位、和2014年获得美国北卡罗来纳大学生物信息学博士学位。主持中国国家自然科学基金青年和面上项目,参与科技部“十四五”重点研发计划、美国国立卫生研究院基础研究课题U01、R01项目、中国汤臣倍健和瑞士雀巢健康研究院横向合作项目。已发表领域内高影响的SCI论文共计40篇,其中通讯/第一作者发表SCI论文16篇。
● 更多信息,请参考课题组网页(包含博士后招聘信息):http://nilab.met-bioinformatics.cn/
更多推荐
(▼ 点击跳转)
iMeta文章中文翻译+视频解读
iMeta | 中科院生态中心邓晔组发布微生物组网络分析平台iNAP
▸▸▸▸
iMeta | 南科大宋毅组综述逆境胁迫下植物向微生物组求救的遗传基础(附招聘)
▸▸▸▸
iMeta:青岛大学苏晓泉组开发跨平台可交互的微生物组分析套件PMS

▸▸▸▸
iMeta:德布鲁因图在微生物组研究中的应用
▸▸▸▸
iMeta:哈佛刘洋彧等基于物种组合预测菌群结构的深度学习方法
▸▸▸▸
iMeta:吴青龙/王明福/刘金鑫等-从肠道菌群看待人类对高原饮食的适应性
▸▸▸▸
iMeta:西农韦革宏团队焦硕等-土壤真菌驱动细菌群落的构建
▸▸▸▸
iMeta:高颜值高被引绘图网站imageGP
iMeta教你绘图
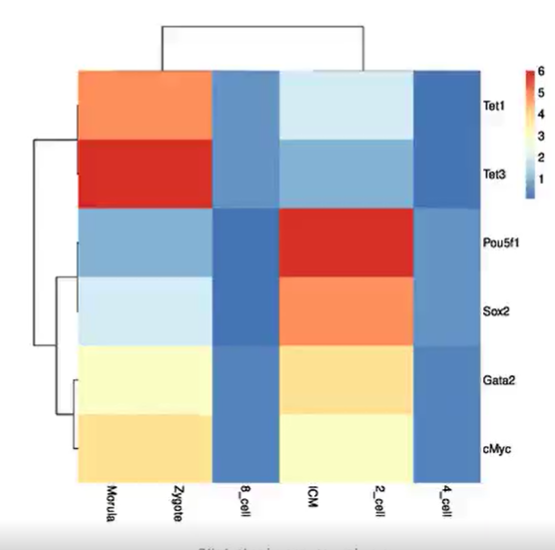
使用ImageGP绘图热图Heatmap
▸▸▸▸
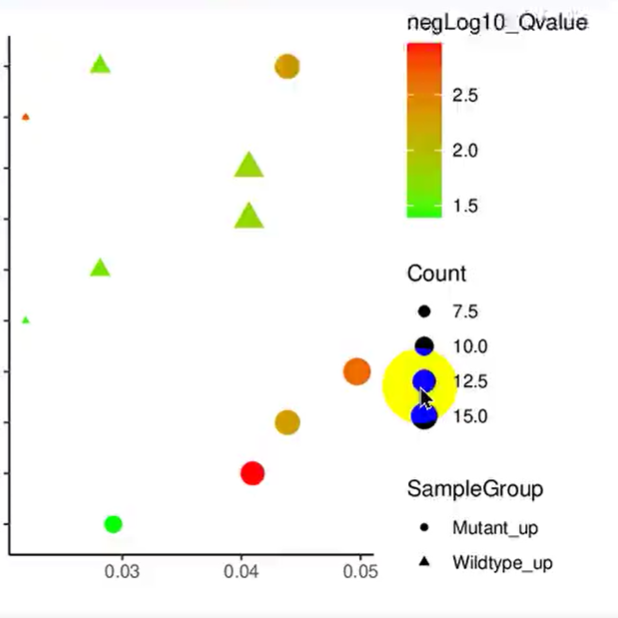
使用ImageGP绘图富集分析泡泡图
期刊简介

“iMeta” 是由威立、肠菌分会和本领域数百位华人科学家合作出版的开放获取期刊,主编由中科院微生物所刘双江研究员和荷兰格罗宁根大学傅静远教授担任。目的是发表原创研究、方法和综述以促进宏基因组学、微生物组和生物信息学发展。目标是发表前10%(IF > 15)的高影响力论文。期刊特色包括视频投稿、可重复分析、图片打磨、青年编委、前3年免出版费、50万用户的社交媒体宣传等。2022年2月正式创刊发行!
联系我们
出版社:https://onlinelibrary.wiley.com/journal/x
投稿:https://mc.manuscriptcentral.com/imeta
邮箱:

微信公众号
iMeta
责任编辑
微微
往期精品(点击图片直达文字对应教程)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|




























后台回复“生信宝典福利第一波”或点击阅读原文获取教程合集


版权声明:本文内容由互联网用户自发贡献,该文观点仅代表作者本人。本站仅提供信息存储空间服务,不拥有所有权,不承担相关法律责任。如发现本站有涉嫌侵权/违法违规的内容,请联系我们,一经查实,本站将立刻删除。
如需转载请保留出处:https://51itzy.com/kjqy/54395.html