
1)测序质量值
首先在了解fastq,fasta之前,了解一下什么是质量值。Phred 功能是处理测序仪直接生成的色谱图,给出相应的碱基和质量值。不同的测序仪会给出不同的色谱文件,Phred 能够识别三种格式的色谱文件,SCF, ABI 和预先处理的 ESD 格式。 碱基的测序质量值 Q 和此碱基出错的概率 Pe 相关。公式:Q = -10 log10( Pe )。phred软件在对reads进行base calling的时候会给出每一个碱基的质量值,这个质量值的计算与测序预期错误率相关(estimated probability of error):
Phred Quality Score Probability of incorrect base call Base call accuracy 10 1 in 10 90 % 20 1 in 100 99 % 30 1 in 1000 99.9 % 40 1 in 10000 99.99 % 50 1 in 99.999 % 讯享网
除此之外还有solexa标准,即将p换成了p/(1-p),其他完全按照sanger的定义来做。当测序质量很高的情况下两种形式几乎没区别,但低质量的碱基则有区别了(如图)
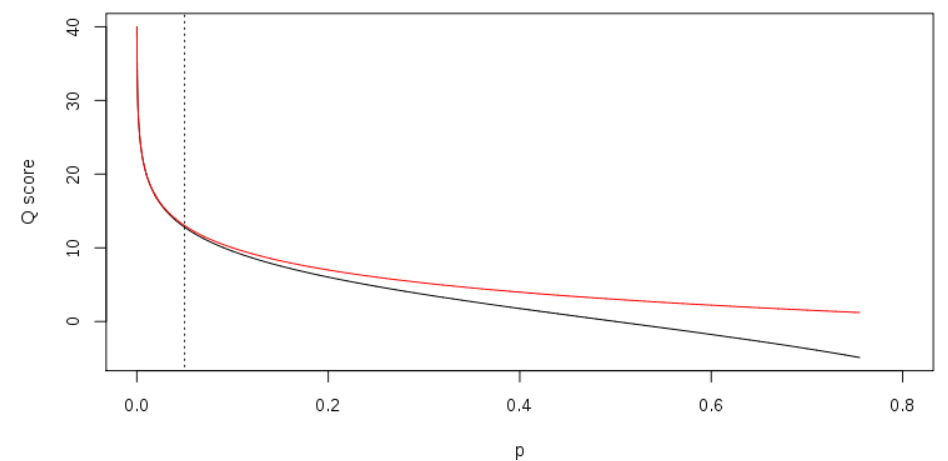
Qscore与p之间的关系,其中红线表示Q=-10 log10p标准,黑色实线表示Q=-10 log10p/(1-p)标准。
1.1)ACII码
为了方便储存及可读这些信息,利用可打印的ACII码将这些质量值转化为单字符single characters (or bytes)。ASCII 字符集,最基本的包含了128 个字符。其中前 32 个, 0-31 ,即 0x00-0x1F ,都是不可见字符,这些字符,为控制字符。可见字符为32–126。sanger-fastaq格式用 ASCII 33–126 来表示phred 质量值 0 到93 。举例来说:一般地,碱基质量从0-40,既ASCii码为从 “!”(0+33)到“I”(40+33)。如果某碱基测序出错的概率为0.001,则Q应该为30。则30+33=63,那么63对应的ASCii码为“?”,在第四行中该碱基对应的质量代表值即为“?”。

2)fastq格式
fastq格式是一个文本格式用于贮存生物学序列及其相应质量值(通常是核酸序列的)。fastq格式是一种包含质量值的序列文件,其中的q为quality,一般用来存储原始测序数据,扩展名一般为fastq或者fq。目前illumina测序,BGISEQ,Ion Torrent,pacbio,nanopore都以fastq格式存储测序数据,其中illumina,BGISEQ一般是双末端测序,一般是一对文件,命名为*_R1.fq.gz与*_R2.fq.gz。为了简洁,这些序列以及质量信息使用ASCII字符标示。该格式最初由Sanger开发,目的是将FASTA序列与质量数据放到一起,目前已经成为高通量测序结果的事实标准。通常fastq文件中每一个序列含有4行信息(如下):

第一行:以‘@’开头,是这一条read的名字,这个字符串是根据测序时的状态信息转换过来的,中间不会有空格,它是每一条read的唯一标识符,同一份FASTQ文件中不会重复出现,甚至不同的FASTQ文件里也不会有重复;
第二行:测序read的序列,由A,C,G,T和N这五种字母构成,这也是我们真正关心的DNA序列,N代表的是测序时那些无法被识别出来的碱基;
第三行:以‘+’开头,在旧版的FASTQ文件中会直接重复第一行的信息,但现在一般什么也不加(节省存储空间);
第四行:测序read的质量值,每个字符与第二行的碱基一一对应,按照一定规则转换为碱基质量得分,进而反映该碱基的错误率,因此字符数必须和第二行保持一致。对于每个碱基的质量编码标示,不同的软件采用不同的方案 。目前有5种:
讯享网1、Sanger,Phred quality score:值的范围从0到93,对应的ASCII码从33到126,但是对于测序数据(raw read data)质量得分通常小于60,序列拼接或者mapping可能用到更大的分数。
2、Solexa/Illumina 1.0, Solexa/Illumina quality score:值的范围从-5到62,对应的ASCII码从59到126,对于测序数据,得分一般在-5到40之间;
3、Illumina 1.3+,Phred quality score:值的范围从0到62对应的ASCII码从64到126,低于测序数据,得分在0到40之间;
4、Illumina 1.5+,Phred quality score:但是0到2作为另外的标示,详见http://solexaqa.sourceforge.net/questions.htm#illumina
5、Illumina 1.8+
不同的标准之间可以相互转化换。注意:第二行@字符,第三行+字符,在第四行质量值中会出现,有时也会在行首出现,因此在处理fastq格式的时候要格外的关注。
2.1)如何打开fq文件呢?
有多种方式可以打开fq文件: 1. 最为原始的是在Linux服务器上用查看文本的less命令或cat命令;2.同时也可以用pycharm软件打开fq文件,直接拖入打开即可;
2.2)获取fastq文件
将样本测序,就可以得到fastq格式文件,无论人,动物,植物,微生物,测序的是全基因组,还是外显子,捕获序列,抑或是RNA样本,FFPE样本最终得到的都是fatsq格式文件。注意,有些平台得到的是两个文件,reads1与reads2,有些平台得到的是一个文件。也可以在NCBI SRA数据库下载测序文件,使用sratools工具中的prefetch或者fastq-dump软件都可以下载fastq文件。
2.3)fastq文件统计
如果想对fastq文件进行统计,例如统计序列条数,碱基总数,reads读长分布等,可以使用seqkit工具进行操作。
如果想统计fastq文件每条序列ATCG四种碱基组成以及质量值分布,可以使用seqtk comp工具来完成。
2.4)过滤短的序列
Ion Torrent,pacbio,nanopore测序的fastq文件序列长度并不相同,通常需要过滤较短的序列,例如过滤掉长度小于150bp的序列。可以使用seqtk seq或者seqkit seq进行操作。
2.5)转换为列表格式
如何将fastq格式转换为列表格式?可以使用seqkit fx2tb,为什么要做这一步处理呢,转换为列表,这样方便根据ID进行处理。将四行数据转换为一行三列,这样就可以使用常用的列表处理程序来进行处理,例如awk。当然处理完了,还可以使用tab2fx将列表转为换fastq格式。
2.6)质量值转换

目前测序得到的fastq文件,都采用phred+33的格式,但是如果处理之前的文件,还有可能遇见phred+64的模式,一般软件中包含--phred33或者--phred64选项,当然也可以直接在两种质量值之间进行转换。
2.7)去除接头adapter
接头adapter主要是指illumina测序时加入的P7接头与P5接头,理论上来说测序从测序引物开始,是测序不到接头的,但是由于部分打断片段果断或者由于adapter空载,会导致adpter自身连接,以上两种情况都会导致测序reads中包含adapter序列。adapter序列非基因组本身的序列,会干扰分析,因此需要去除掉。主要是illumina测序中包含adapter。去除adapter主要是采用两种方式,一种是直接给定adapter序列,与reads比对,比对上的就把整条reads去掉,另外一种是测序完成之后,给定一个adater list文件,其中包含了含有adapter序列的reads ID列表,给定一个阈值将这些reads去除掉。cutadapt可以根据给定的adapter序列进行过滤。
2.8)去除低质量
低质量主要是指去除测序质量错误率较高的位点,一般以Q20作为标准,如果一个碱基质量值低于Q20,则认为一个低质量,如果一条序列中低质量碱基达到一定比例,例如达到40%以上,则过滤掉此条序列。是过滤数据主要处理指标。seqtk工具seq功能通过-q,-n可以将低质量碱基进行标记,例如替换为小写字母或者其他字符,但是不进行过滤,有专门的数据过滤工具。
2.9)数据过滤
有很多软件可以一次性完成数据过滤工作,包括去除adapter,去除低质量,去除N碱基,去除duplication,截取reads,常用的包括fastp,trimmomatic,SOAPnuke等,不过这些软件都各有优缺点,SOAPnuke很好用,但是去除adapter需要提供一个adapter reads ID的列表,从网上下载的数据没有这个,fastp利用固定adapter序列,但是不能去除duplication,trimmomatic选项参数太长,而且也不是很好用。这里推荐使用fastp。
2.10)排序和抽样
如果想对fastq格式文件进行排序,可以使用seqkit sort功能。有时候需要从全部文件中抽取一部分进行分析,因为测序出来的数据本身就是随机分布的,因此,即使从头到尾开始取数据,出来的也是随机的。当然还是可以随机抽样的。seqtk和seqkit工具都提供了抽样功能。
2.11)拆分数据
有时候需要将fastq文件拆成多份,或者根据固定模式进行拆分,例如测序时同一个lane的数据根据index进行拆分,16S测序中,同一个文件中序列根据barconde进行拆分等。seqtk split与split2可以用来拆分文件,既可以按照大小进行拆分,也可以直接拆分成固定份数。
3)fasta格式
3.1)fasta格式最初来自FASTA软件包,也是一种文本格式,以单字符( single-letter codes)贮存核酸或者蛋白序列信息,允许在序列前加注释信息。由2部分信息组成:
>gi||gb|AAD44166.1| cytochrome b [Elephas maximus maximus]
LCLYTHIGRNIYYGSYLYSETWNTGIMLLLITMATAFMGYVLPWGQMSFWGATVITNLFSAIPYIGTNLVEWIWGGFSVDKATLNRFFAFHFILPFTMVALAGVHLTFLHETGSNNPLGLTSDSDKIPFHPYYTIKDFLGLLILILLLLLLALLSPDMLGDPDNHMPADPLNTPLHIKPEWYFLFAYAILRSVPNKLGGVLALFLSIVIGLMPFLHTSKHRSMMLRPLSQALFWTL***LLTLTWIGSQPVEYPYTIIGQMASILYFSIILAFLPIAGXIENY
3.2)fasta格式在拓展的文件命名中,一般会约定俗成,具体见下表格:

我们通常进行处理的文件是fasta文件,如何将fastaq无损的转化成fasta文件? 有以下几种方式可以将fq格式文件转化成fasta文件:
在Linux服务器上用基础命令来实现这个转换:
zless -S SRR_1.fastq.gz| paste - - - -|cut -f 1-2|tr '\t' '\n'|tr '@' '>' |less -S > SRR_1.fasta #结果为: >SRR.1 HWI-ST177:290:C0TECACXX:1:1101:1373:2104 length=63 TGGGAGGCTGAGGCAGGAGAATCACTTAAACCTGGGAGGCAGAGGTTACAGTGAGCCGAGATT >SRR.2 HWI-ST177:290:C0TECACXX:1:1101:1340:2124 length=63 AAAGAAGGCGACAGTGAGAAGGAGTCCGAGAAGAGTGATGGAGACCCAATAGTCGATCCTGAG >SRR.3 HWI-ST177:290:C0TECACXX:1:1101:1273:2183 length=63 CTGCTGGGCCCCAAGGTCCTCCTGGTCCCAGTGGTGAAGAAGGAAAGAGAGGCCCTAATGGGG >SRR.4 HWI-ST177:290:C0TECACXX:1:1101:1562:2147 length=63 CTTGGCTGCAGCCATCCCGCTTAGCCTGCCTCACCCACACCCGTGTGGTACCTTCAGCCCTGG # 同时生成SRR_1.fasta文件。 # 同时zcat SRR_1.fastq.gz| paste - - - -|cut -f 1-2|tr '\t' '\n'|tr '@' '>' |less -S 也可实现这种转换如果你不需要查看fq中的具体内容,可以用软件实现fq到fasta文件的转换,fastX_tool软件可以实现相同的功能
讯享网fastq_to_fasta -Q33 -v -n -i SRR_1.fastq -o SRR_1.fasta #其中-Q33是必须添加的参数
一些软件只支持fasta格式,例如只有fasta格式才能进行blast比对,因此有时候如果需要将测序数据直接进行blast比对,就需要将fastq转为换fasta,将fastq转换为fasta有多种方法,例如seq,awk,perl都可以,还有很多单独的程序。这里使用seqtk和seqkit分别演示一下。
#seqtk工具 seqtk seq -A nanopore.fastq.gz #seqkit工具 seqkit fq2fa nanopore.fastq.gz
版权声明:本文内容由互联网用户自发贡献,该文观点仅代表作者本人。本站仅提供信息存储空间服务,不拥有所有权,不承担相关法律责任。如发现本站有涉嫌侵权/违法违规的内容,请联系我们,一经查实,本站将立刻删除。
如需转载请保留出处:https://51itzy.com/kjqy/42342.html