
PHIAF: prediction of phage-host interactions with GAN-based data augmentation and sequence-based feature fusion
期刊名:Briefings in Bioinformatics
IF: 13.994
中科院分区:一区
代码链接: https://github.com/BioMedicalBigDataMiningLab/PHIAF
出版日期:2021年9月1日
Abstract
噬菌体疗法已成为抗生素治疗细菌性疾病最有希望的替代方案之一,鉴定噬菌体-宿主相互作用(PHI)有助于了解噬菌体感染细菌的可能机制,从而指导噬菌体疗法的发展。在本文中,我们提出了一种 PHI 预测方法,该方法基于生成对抗网络 (GAN) 的数据增强和基于序列的特征融合 (PHIAF)。
Model architecture
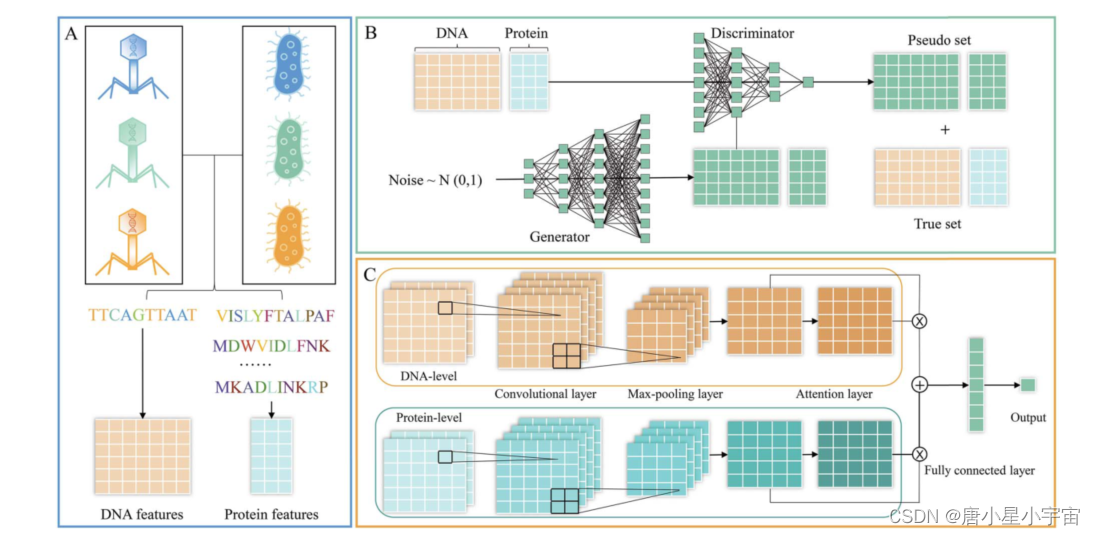
Materials and methods
Dataset
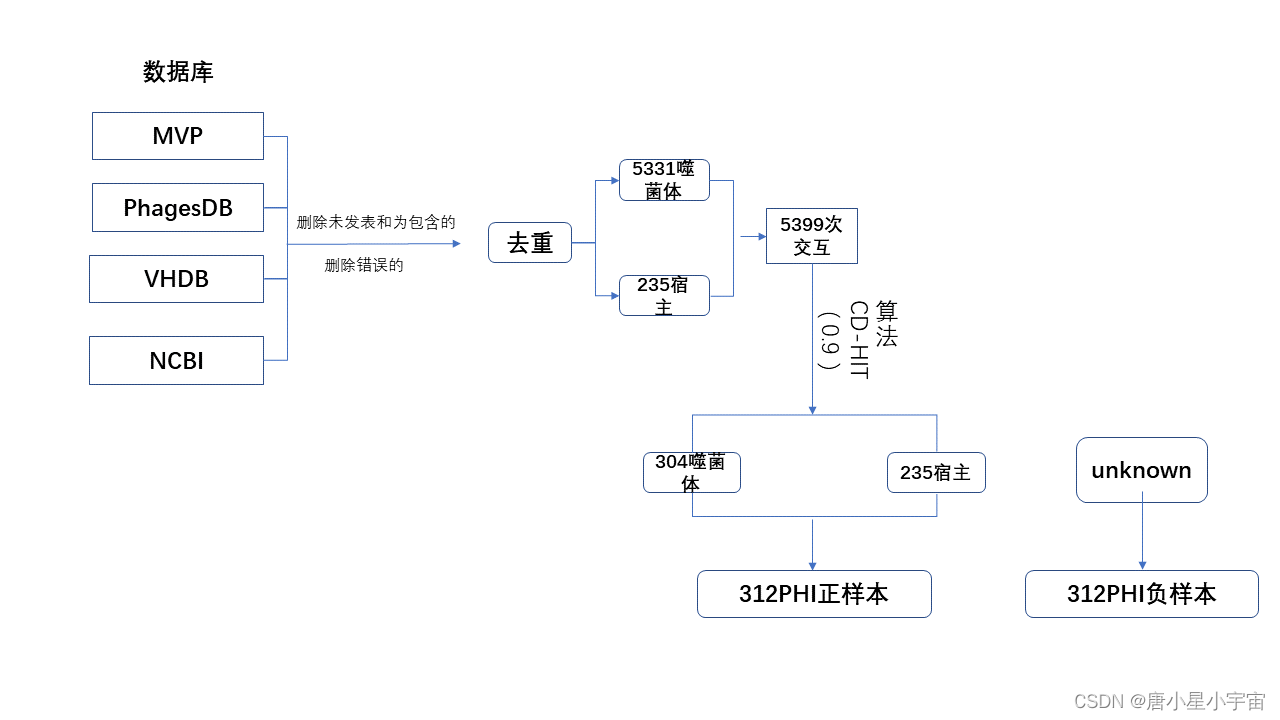
本文从四个广泛使用的数据库下载数据(包括噬菌体、宿主及其相互作用),包括MVP、PhagesDB、VHDB、NCBI合并这些数据以构建具有更多 PHI 的数据集以供研究。首先,删除未在文献中发表或未包含在 NCBI 记录中的 PHI,以确保 PHI 可靠。其次,根据噬菌体的定义删除错误标记为噬菌体/宿主的数据,删除的数据包括不属于病毒的噬菌体和不属于细菌或古细菌的宿主。第三,从 NCBI 数据库中提取剩余噬菌体和宿主的全基因组序列和编码蛋白序列。我们将四个数据库中剩余的噬菌体和宿主结合起来,去除重复,得到5331个噬菌体和235个宿主之间的共5399次交互。噬菌体的数量远大于宿主的数量,一个宿主可能与多个噬菌体相互作用。我们使用算法 去除每个宿主具有高相似性的冗余噬菌体。我们将 0.90 设置为高相似度阈值,这与 CD-HIT 工具 的默认阈值相同。减少冗余后,获得了一个基准数据集,其中包含 304 个噬菌体和 235 个宿主之间的 312 次交互,可用于更好地评估预测模型的性能。在这个数据集中,我们将 312 个已知的 PHI 设置为正样本,并从所有未知的 PHI 中选择负样本,同时确保正样本和负样本的数量相等。
Feature extraction module

由于每个噬菌体/宿主都有多个蛋白质序列,我们考虑六个算子(平均值、最大值、最小值、标准差、方差和中值)来整合蛋白质序列的特征,并为每个噬菌体/宿主获得一个 162 维的特征向量。
Data augmentation module
对抗生成网络(Generative Adversarial Network, GAN)
GAN包括两个模型组成部分,其一是(Generative model),其二是(Discriminative model),简单来说生成器的作用就是生成伪正样本数据,判别器的作用是来判断输入的数据是正样本,还是伪造的正样本。大致框架如下:
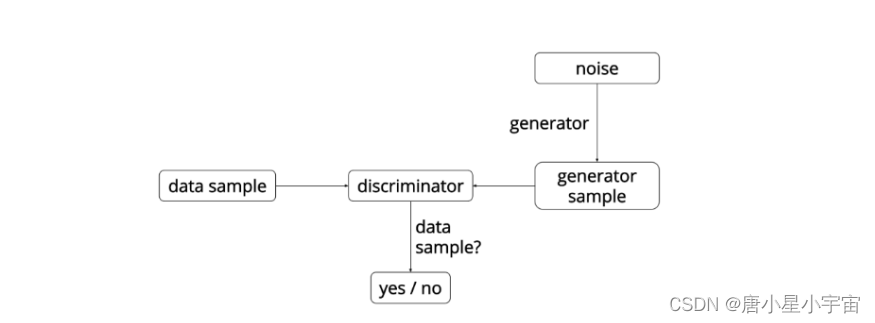
在训练过程中,生成网络的目标就是尽量生成真实的数据(伪正样本)去欺骗判别网络D。而判别网络D的目标就是尽量把网络G生成的数据和真实的数据分别开来。这样,G和D构成了一个动态的“博弈过程”。
本文使用GAN的主要目的在于: 解决模型训练数据集中PHI的数据稀疏性
class Generator(nn.Module): def __init__(self,shape1): super(Generator, self).__init__() main = nn.Sequential( nn.Linear(shape1, 128), nn.ReLU(True), nn.Linear(128, 256), nn.ReLU(True), nn.Linear(256, 512), nn.ReLU(True), nn.Linear(512, 1024), nn.Tanh(), nn.Linear(1024, shape1), ) self.main = main def forward(self, noise, real_data): if FIXED_GENERATOR: return noise + real_data else: output = self.main(noise) return output class Discriminator(nn.Module): def __init__(self,shape1): super(Discriminator, self).__init__() self.fc1=nn.Linear(shape1, 512) self.relu=nn.LeakyReLU(0.2) self.fc2=nn.Linear(512, 256) self.relu=nn.LeakyReLU(0.2) self.fc3 = nn.Linear(256, 128) self.relu = nn.LeakyReLU(0.2) self.fc4 = nn.Linear(128, 1) def forward(self, inputs): out=self.fc1(inputs) out=self.relu(out) out=self.fc2(out) out=self.relu(out) out=self.fc3(out) out=self.relu(out) out=self.fc4(out) return out.view(-1) 讯享网
PHI prediction module
基于增强数据集,本文使用噬菌体和宿主的 DNA 和蛋白质序列衍生特征来构建预测模型。由于序列衍生特征通常具有复杂的短程/长程依赖性,本文将 DNA/蛋白质特征向量重塑为“图像”,以捕捉它们维度之间的复杂关系,我们首先采用 Min-Max 归一化将特征向量中的值归一化到 0 到 1 的范围内。令 N 表示 DNA/蛋白质的特征向量的维数;然后,通过按行放置值将特征向量重塑为 n × n“图像”,其中 n 满足条件:(n - 1) × (n - 1) < N 和 N ≤ n × n。当 n N < n × n 时,我们通过向剩余的 n × n - N 个条目添加零来实现填充。最后,获得了每个噬菌体(或宿主)的 DNA 和蛋白质序列衍生的特征矩阵。在模型中引入了注意力机制,添加了注意力层来捕获重要特征,然后整合这些特征。在注意力层中,将 DNA 和蛋白质级别的特征图(Od 和 Op)输入全连接层,分别计算权重相量(αd 和 αp);然后,特征图乘以相应的权重向量。注意力层的输出计算如下:

最后,注意力层的输出被输入到一个两层的 MLP 中,以产生样本是 PHI 的概率。

讯享网def attention_3d_block(inputs): input_dim = int(inputs.shape[2]) a = Permute((1, 2))(inputs) a = Dense(32, activation='softmax')(a) a_probs = Permute((1, 2))(a) output_attention_mul = Multiply()([inputs, a_probs]) return output_attention_mul
Comparison with state-of-the-art methods
为了证明 PHIAF 的有效性,我们将其与以下最先进的方法进行比较,包括基于 DNA 序列的算法和基于蛋白质序列的方法。
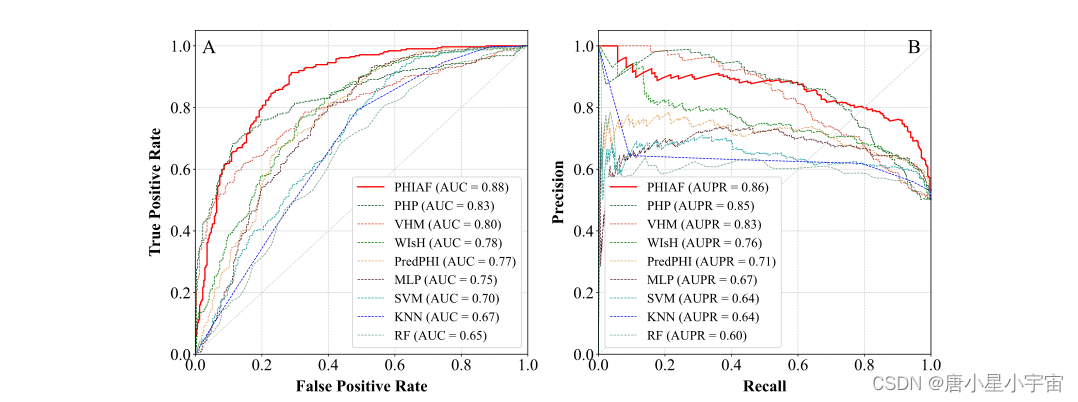
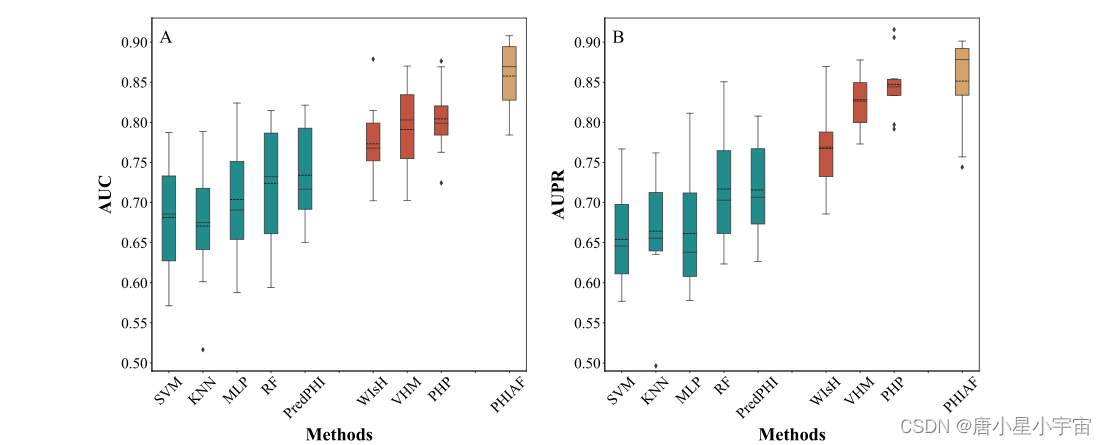
我们随机选择数据集中三分之一的噬菌体和宿主,并将它们及其相互作用用作测试集。剩余的噬菌体和宿主及其相互作用用于训练预测模型。在此实验设置下,测试集中的噬菌体或宿主不包括在训练集,它们被视为看不见的数据。我们基于训练集训练预测模型,然后将训练好的模型应用于测试集,以评估预测模型在未见数据上的性能。为避免评估偏差,我们重复训练和测试 10 次,并采用平均/中值性能。如图 3 所示,PHIAF 优于其他方法,产生更高的平均 AUC (0.86) 和 AUPR 得分 (0.85) 以及合理的标准差,AUC 和 AUPR 得分的中位数分别为 0.88 和 0.87。与 5-CV 结果类似,基于 DNA 序列的方法比基于蛋白质序列的方法产生更好的结果。此外,5-CV 的结果与未见数据测试的比较表明,我们的方法在两种情况下都实现了非常相似的性能(AUC 和 AUPR 相差 2% 和 1%),这些结果表明我们提出的方法是稳健的并且可以在看不见的数据上表现良好,这表明 PHIAF 是一种很有前途的工具,可以从序列数据中识别 PHI。
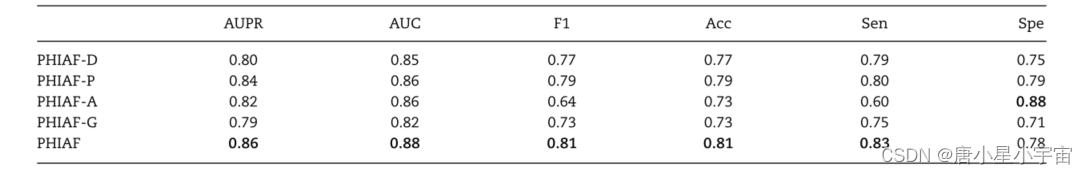
Ablation study
PHIAF-D: 不使用 DNA-level features.
PHIAF-P: 不使用 protein-level features.
PHIAF-A: 不使用 attention layer.
PHIAF-G: 不使用 pseudo samples
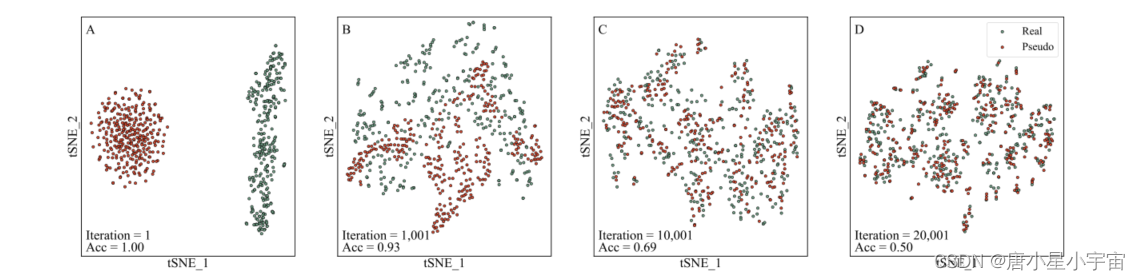
Discussion
为了证明真实样本和伪样本无法区分,并且伪样本可以用作训练正样本,我们使用 t 分布随机邻域嵌入(t-SNE)来可视化真实和伪正样本的 2D 分布GAN 不同训练迭代的样本。
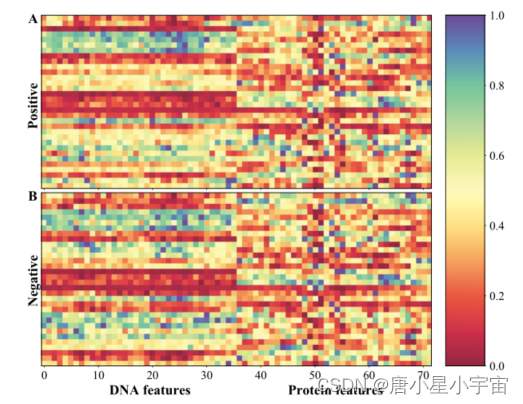
此外,我们分析了分配给不同特征的注意力层中的权重,以研究注意力机制学习到的特征的重要性。
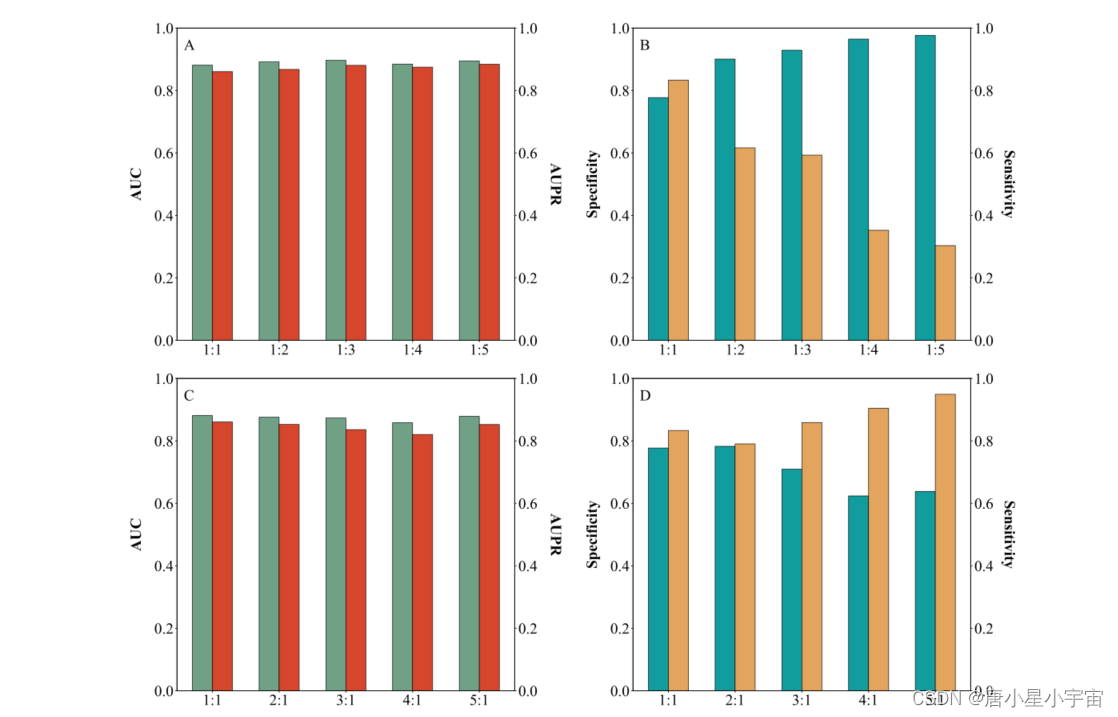
基于具有不同正负样本比例(1:1、1:2、1:3、1:4、1:5、2:1、3:1、4:1 和5:1) 分析负样本或伪正样本的数量如何影响 PHIAF 的性能。
Case study
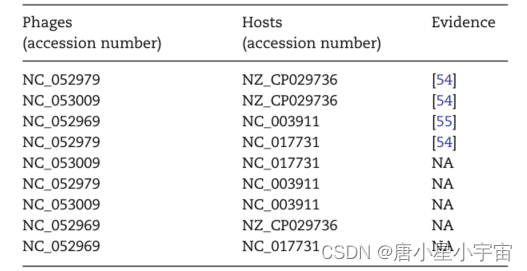
在本节中,我们进行了一个案例研究,以估计 PHIAF 预测未知新 PHI 的能力。首先使用 2021 年 1 月 1 日之前出现在 NCBI 中的所有噬菌体和宿主的已知相互作用来训练 PHIAF,以识别剩余噬菌体和宿主之间的所有对(其余代表 2021 年 1 月 1 日之后出现在 NCBI 中的噬菌体和宿主)。然后,我们根据预测分数对 PHI 进行排序,并搜索新发表的文献,以验证预测的 PHI 是否已被生物实验证实。
Conclusion
抗生素的过度使用给细菌性疾病的治疗带来了一些严峻的挑战。作为治疗细菌性疾病最有希望的抗生素替代品之一,噬菌体疗法受到了广泛关注。在本研究中,我们提出了一种用于 PHI 预测的 PHIAF 方法,该方法利用 GAN 生成高质量的伪样本,融合来自 DNA 和蛋白质序列的特征以获得更好的性能,并使用注意力机制来提供预测模型的可解释性。通过 5-CV 与最先进的方法进行比较表明,PHIAF 实现了** PHI 预测(就 AUC 和 AUPR 而言,性能平均提高了大约 13.64% 和 14.75%)。
版权声明:本文内容由互联网用户自发贡献,该文观点仅代表作者本人。本站仅提供信息存储空间服务,不拥有所有权,不承担相关法律责任。如发现本站有涉嫌侵权/违法违规的内容,请联系我们,一经查实,本站将立刻删除。
如需转载请保留出处:https://51itzy.com/kjqy/118969.html