
变异标准化
简约原则和左对齐
VCF 中变异表示的规范化由两部分组成:简约原则和左对齐。
简约原则(Parsimony):以尽可能少的核苷酸表示变异,且allele 长度不能为0。
例如,GC->GCA 是可以简化为C->CA
左对齐(Left alignment):将变异起始位置向左移动,直到不能再移动为止。
例如,GCACACA -> GCACA 表示的是CA的deletion, 实际上,CA的deletion是可以有三个位置可以选择:
GCACACA、GCACACA、GCACACA; 而左对齐的目标就是把这个del移到最左边。因此简化和左对齐后的结果就变为了GCA -> G
下面也可以以Adrian Tan等[2] 提到的例子简单说说:
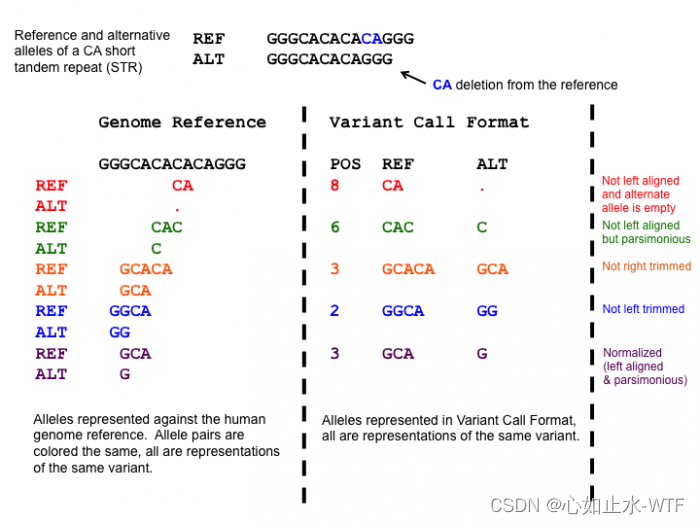
这个例子中的变异是一个CA 的deletion 变异, 出现了图中五种不同的表达方式,其中只有紫色的同时符合简约和左对齐原则。
ALT长度为0, 显然是不合理的
没有左对齐,可以看到,CA是明显可以再往左边移动的2bp(因为CA的左侧ref上也是CA),不过这个变异已经是最简约的形式了
右侧的CA是多余的,可以切除
左侧的G是多余的,可以切除
简约且左对齐
讯享网

变异标准化代码和工具实现
Adrian Tan等实际上在文章中已经给出了变异标准化的伪代码实现逻辑:
简单来说:如果 REF 和 ALT 尾部(最右端)具有相同的碱基则切除相同的碱基; 切除之后,如果存在REF/ALT为空,则向左侧延伸一个碱基,保证REF/ALT不为空(此处是需要有参考基因组的,即fasta文件); 最后,检查REF和ALT左侧是否具有相同碱基且当REF和ALT的长度都大于或等于2(保证切除后不会为空) 时, 切除左侧相同的碱基。最终得到的REF、ALT即为标准化的变异结果了。

目前不少工具都有变异标准化的功能,如gatk, bcftools, vt等
#bcftools bcftools norm -f ref.fa in.vcf -O z > out.vcf.gz #gatk java -jar GenomeAnalysisTK.jar -T LeftAlignAndTrimVariants --trimAlleles -R ref.fa --variant in.vcf.gz -o out.vcf.gz #vt vt normalize -r ref.vcf.gz -o out.vcf.gz 讯享网
github上也有一些简单代码可以实现,例如 https://github.com/wonderful1/variation_normalize
左对齐和右对齐
尽管我们前面一直强调变异左对齐,一般大家也是统一使用左对齐的标准。 实际上右对齐也是存在的,可以参考生信大佬黄树嘉的博客 关于Indel,我该往左还是该往右。 简而言之,当我们使用注释软件对vcf文件进行突变的基因、C点、P点等信息标注时需要按照目前业内共识、临检中心室间质评要求的HGVS规范来实行,而HGVS规范明确规定当序列改变和氨基酸功能一致等价时,突变的命名要以靠近 3-UTR 区来命名。
例如下图两个例子:
1)由于c.17至c.34是一连串AT, 当发现有一个AT缺失时, 可认为发生在该区段的多个位置, 而HGVS矫正后, 正确的命名是c.33_34del。
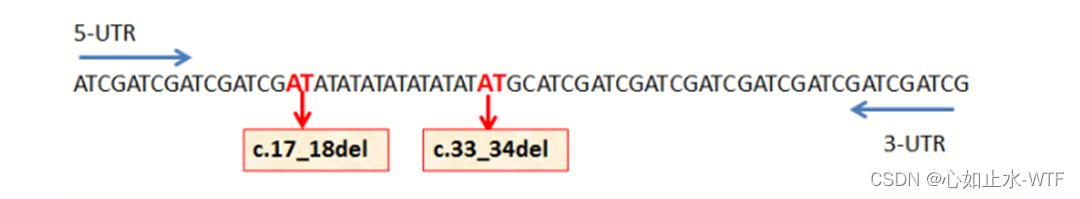
2)由于蓝色框里TCT的两边都是A, 当发生包含TCT的4bp缺失时, 没缺的那个A可认为是c.17也可认为是c.21, 而HGVS矫正后, 正确的命名是c.18_21delTCTA。

右对齐的实现代码可以参考 https://github.com/counsyl/hgvs/blob/master/pyhgvs/variants.py
参考
- Adrian Tan, Gonçalo R. Abecasis, Hyun Min Kang, Unified representation of genetic variants, Bioinformatics, Volume 31, Issue 13, 1 July 2015, Pages 2202–2204, https://doi.org/10.1093/bioinformatics/btv112
- https://genome.sph.umich.edu/wiki/Variant_Normalization
版权声明:本文内容由互联网用户自发贡献,该文观点仅代表作者本人。本站仅提供信息存储空间服务,不拥有所有权,不承担相关法律责任。如发现本站有涉嫌侵权/违法违规的内容,请联系我们,一经查实,本站将立刻删除。
如需转载请保留出处:https://51itzy.com/kjqy/61936.html