
1, 序言
这篇是基因组选择的理论加实践,因为我看到一句话,Talk is cheap. Show me the code,很有感触,有感而写。使用的包是R的sommer和asreml,其实强健的还是成熟的软件,比如DMU,BLUPF90,PIBLUP,ASreml等,但sommer作为基本功能的演示,非常合适。
2, 定义
基因组选择(Genomic Selection, GS), 利用覆盖全基因组的高密度分子遗传标记进行的标记辅助选择.
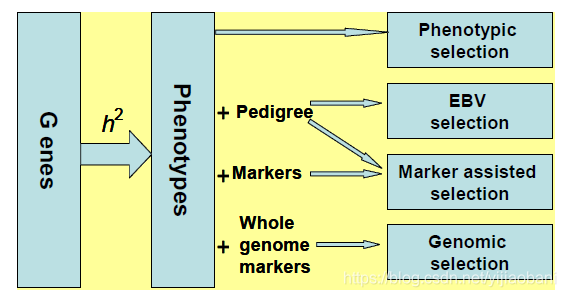
- 表型选择, 这对于遗传力高的性状选择有效, 即表型可以遗传比例较大的性状, 比如植物里面千粒重, 比如动物里面的初生重等.
- EBV selection, 应用系谱的动物模型
- Marker assisted selection(MAS), 分子标记辅助选择, 对于有主效QTL或者主效基因的性状, 有优势.
- Genomic selection, 全基因组选择
选择进展的定义
选 择 进 展 = 选 择 强 度 × 选 择 准 确 性 × 遗 传 标 准 差 世 代 间 隔 选择进展 = \frac{选择强度 \times 选择准确性\times 遗传标准差}{世代间隔} 选择进展=世代间隔选择强度×选择准确性×遗传标准差
- 选择强度: Intensity of selection
- 选择准确性: Accuracy of selection
- 遗传标准差: Genetic standard deviation
- 世代间隔: Generation interval
3, 不同选择方法的比较
基于系谱的动物模型
局限:
- 对低遗传力性状效果较差
- 不容易度量的性状(胴体性状, 肉质性状)效果较差
- 不能早期度量的性状, 效果较差
分子标记辅助育种(MAS)
局限:
- 需要先对主效基因或者QTL进行检测
- 不同群体变化较大
- 标记可解释的遗传变异百分比较低
- 在动物育种中的应用非常有限
全基因组选择
优点:
- 无需进行主效基因或者QTL的检测
- 不依赖于表型信息(候选群)
- 能够捕获基因组中的全部变异
- 对于低遗传力, 难以度量的性状提升效果明显
4, 基因组选择流程
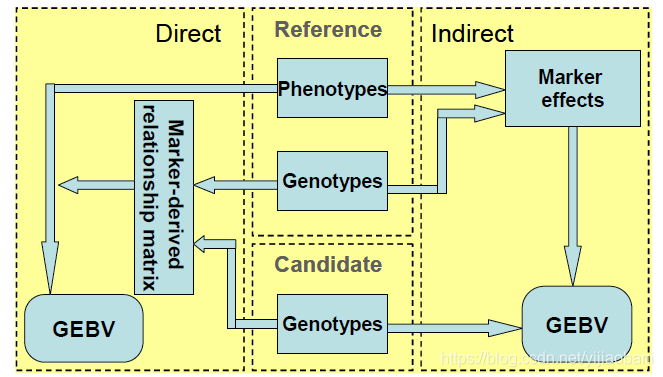
- 1, 建立参考群体
- 获得每个个体的性状表型值
- 测定每个个体的SNP基因型(使用SNP芯片)
- 估计SNP效应值
- 2, 在候选群体中进行基因组选择
- 测定候选个体的SNP基因型
- 计算个体的GEBV
- 依据GEBV进行选择
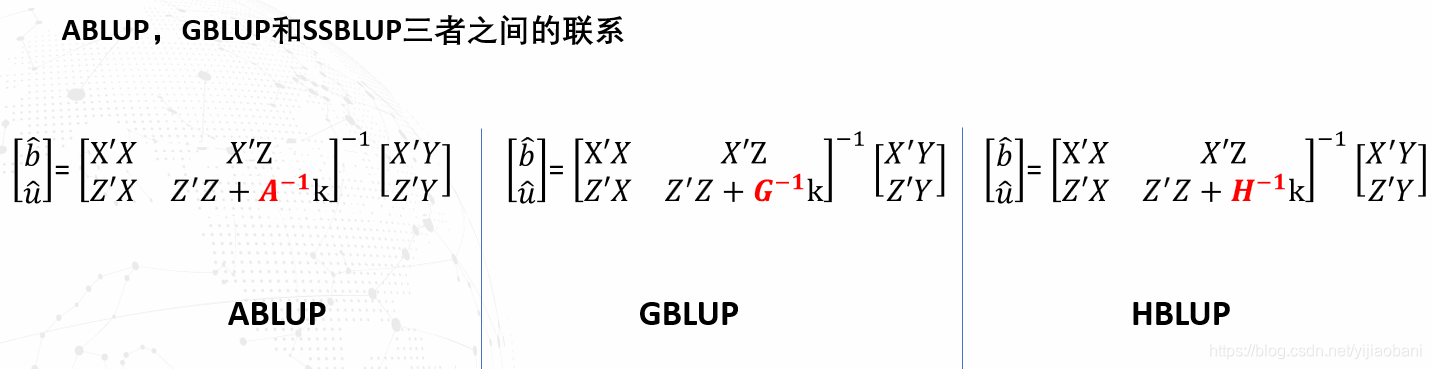
动物模型, GBLUP方法和Single-setp方法

三种方法的区别
- 动物模型是利用的系谱构建的A矩阵
- GBLUP是利用基因组信息构建的G矩阵
- 一步法(single-setp)是利用系谱和基因组信息构建的H矩阵

5,其它方法
除了GBLUP和Single-step, 还有其它方法用于基因组选择, 不过应用最广最强建最有优势的还是GBLUP和Single-step.
- RR-BLUP(Rirdge Regression BLUP)
- Bayesian method
- Bayes A
- Bayes B
- Bayes C π \pi π
- Bayesian Lasso
- Principal component regression
- Semiparametric procedures
- Machine learning methods
6,Talk is cheap, show me the code
别说话, 上代码

数据描述
- 数据使用QMSim软件模拟而成
- 共有2000个体
- 1200个有基因型
- 共有5个世代, 3,4为参考群, 5为候选群, 每个世代400个体
- 系谱数据为50k
- 遗传力为0.3
软件使用
- QMSim模拟数据
- R包: learnasreml
- R包: sommer
- R包:asreml
# 载入软件包 if (!requireNamespace("devtools")) install.packages("devtools") library(devtools) if (!requireNamespace("learnasreml")) install_github("dengfei2013/learnasreml") library(learnasreml) if (!requireNamespace("sommer")) install.packages("sommer") library(sommer) # 载入数据 data("gs_geno") data("gs_phe") # 动物模型animal model gphe = gs_phe[gs_phe$G %in%c(3:5), ] # 这里提取测序个体 gphe$Progeny = as.factor(as.character(gphe$Progeny)) # 将Progeny转化为因子 ainv = asreml.Ainverse(gphe[,1:3])$ginv # 计算A逆矩阵 moda_as = asreml(Phen ~ Sex + G, random = ~ ped(Progeny),ginverse= list(Progeny=ainv),data = gphe) # 构建模型 summary(moda_as)$varcomp #结果 # GBLUP sommer的操作代码 gdat = gs_geno # 重命名数据 gdat[1:10,1:10] # 预览数据 G = A.mat(gdat-1) # sommer计算G矩阵时, 支持的是-1,0,1, 所以需要转化 time_sommer = system.time({ modg_mm = mmer2(Phen ~ Sex + G, random = ~ g(Progeny),G = list(Progeny=G),data = gphe) # 构建模型 }) # GBLUP asreml的操作代码 library(asreml) diag(G) = diag(G)+0.01 # 矩阵奇异, 对角线+0.01防止奇异 ginv = write_relation_matrix(G,type="ginv") # 转化为三元组形式 attr(ginv,"rowNames") = rownames(G) # 对ginv进行rowNames的设置 time_asreml = system.time({ modg_as = asreml(Phen ~ Sex + G, random = ~ giv(Progeny),ginverse= list(Progeny=ginv),data = gphe,workspace=1e9) # 构建模型 }) # 比较sommer和asreml结果 summary(modg_mm) summary(modg_as)$varcomp # 比较运行时间 time_sommer time_asreml # 计算准确性:asreml blup_as = as.data.frame(coef(modg_as)$random) re1 = cbind(gphe,blup_as) cor(re1$Polygene,re1$effect) # 计算准确性:sommer blup_mm = as.data.frame(randef(modg_mm)) head(blup_mm) names(blup_mm) = "blup_mm" re2 = cbind(gphe,blup_mm) cor(re1$Polygene,re2$blup_mm) 讯享网
方差组分结果比较
讯享网> # 比较sommer和asreml结果 > summary(modg_mm)$var.comp.table VarComp VarCompSE Zratio g(Progeny).Phen-Phen 0. 0.0 4. units.Phen-Phen 0. 0.0 18. > summary(modg_as)$varcomp gamma component std.error z.ratio constraint giv(Progeny).giv 0. 0. 0.0 4. Positive R!variance 1.0000000 0. 0.0 18. Positive
可以看出, sommer和asreml方差组分结果基本一致, Vg=0.194, Ve=0.805
运行时间比较
> # 比较运行时间 > time_sommer 用户 系统 流逝 33.53 3.88 9.47 > time_asreml 用户 系统 流逝 19.00 3.90 23.03 可以看到,sommer运行时间很快,sommer支持的是G矩阵类型,如果使用pedigree,有点麻烦。asreml-r版慢一点,个人体会,asreml-w更快一点。
准确性比较
讯享网> # 计算准确性:asreml > blup_as = as.data.frame(coef(modg_as)$random) > re1 = cbind(gphe,blup_as) > cor(re1$Polygene,re1$effect) [1] 0. > > # 计算准确性:sommer > blup_mm = as.data.frame(randef(modg_mm)) Returning object of class 'list' where each element correspond to one random effect. > names(blup_mm) = "blup_mm" > re2 = cbind(gphe,blup_mm) > cor(re1$Polygene,re2$blup_mm) [1] 0.
因为这是模拟数据,有真值(True breeding value),如果是真实数据,需要用交叉验证的手段评价准确性。
7,后续更新,欢迎持续关注微信公众号
- 1,H矩阵构建及一步法计算
- 2,H矩阵构建不同参数的设置
- 3,多性状GBLUP及多性状一步法计算
- 4,如何进行交叉验证
- 5,如何筛选以及填充基因组数据
- 6,基于贝叶斯的全基因组选择
- 7,基于一步法的GWAS分析
公众号ID:R-breeding

8, 参考资料
张勤老师: 动物基因组选择-理论与应用
Meuwissen T H , Hayes B J , Goddard M E . Prediction of total genetic value using genome-wide dense marker maps[J]. Genetics, 2001, 157(4):1819-29.
Vanraden P M. Efficient methods to compute genomic predictions[J]. Journal of Dairy Science, 2008, 91(11):4414-4423.
Legarra A, Aguilar I, Misztal I. A relationship matrix including full pedigree and genomic information[J]. Journal of Dairy Science, 2009, 92(9):4656-4663.
Aguilar I, Misztal I, Johnson D L, et al. Hot topic: a unified approach to utilize phenotypic, full pedigree, and genomic information for genetic evaluation of Holstein final score.[J]. Journal of Dairy Science, 2010, 93(2):743-752.
Ishizaka A, Labib A. Review of the main developments in the analytic hierarchy process[J]. Expert Systems with Applications, 2011, 38(11):14336-14345.
版权声明:本文内容由互联网用户自发贡献,该文观点仅代表作者本人。本站仅提供信息存储空间服务,不拥有所有权,不承担相关法律责任。如发现本站有涉嫌侵权/违法违规的内容,请联系我们,一经查实,本站将立刻删除。
如需转载请保留出处:https://51itzy.com/kjqy/50407.html