
本文是Seurat包的学习笔记,相比以前的略有更新,重新整理。
2019年7月的生物信息学人才论坛会议上,伊现富老师说过一句话:“过去的流程使用的是过去的工具”,这次重新学单细胞,对这句话有了更深刻的理解。
是的,现在去看曾老板的视频还有豆豆之前的代码(2018-19年的),就有很多跑不通了。
并不是他们写错了,只是工具更新了,原来的函数和数据使用方法变了。
与时俱进这个词,在单细胞的领域体现的淋漓尽致,只有不断学习和进步,才能跟得上这变化的速度。
1.数据和R包准备
代码:https://satijalab.org/seurat/v3.0/pbmc3k_tutorial.html
数据:https://s3-us-west-2.amazonaws.com/10x.files/samples/cell/pbmc3k/pbmc3k_filtered_gene_bc_matrices.tar.gz
rm(list = ls()) options(stringsAsFactors = F) library(dplyr) library(Seurat) library(patchwork) 讯享网
2.读取数据
10X的输入数据是固定的三个文件,在工作目录下新建01_data/,把三个文件放进去。
讯享网dir("01_data/")
[1] "barcodes.tsv" "genes.tsv" "matrix.mtx" 讯享网pbmc.data <- Read10X(data.dir = "01_data/") pbmc <- CreateSeuratObject(counts = pbmc.data, project = "pbmc3k", min.cells = 3, min.features = 200)
Warning: Feature names cannot have underscores ('_'), replacing with dashes ('-') 单细胞相关的这几个R包都是打包成对象
讯享网pbmc
An object of class Seurat 13714 features across 2700 samples within 1 assay Active assay: RNA (13714 features, 0 variable features) 讯享网pbmc.data[c("CD3D","TCL1A","MS4A1"), 1:30]
3 x 30 sparse Matrix of class "dgCMatrix" 讯享网 [[ suppressing 30 column names 'AAACATACAACCAC-1', 'AAACATTGAGCTAC-1', 'AAACATTGATCAGC-1' ... ]]
CD3D 4 . 10 . . 1 2 3 1 . . 2 7 1 . . 1 3 . 2 3 . . . . . 3 4 1 5 TCL1A . . . . . . . . 1 . . . . . . . . . . . . 1 . . . . . . . . MS4A1 . 6 . . . . . . 1 1 1 . . . . . . . . . 36 1 2 . . 2 . . . . 查看表达矩阵,很大一个。
讯享网exp = pbmc[["RNA"]]@counts;dim(exp)
[1] 13714 2700 讯享网 [1] 13714 2700 exp[30:34,1:4]
5 x 4 sparse Matrix of class "dgCMatrix" AAACATACAACCAC-1 AAACATTGAGCTAC-1 AAACATTGATCAGC-1 AAACCGTGCTTCCG-1 MRPL20 1 . 1 . ATAD3C . . . . ATAD3B . . . . ATAD3A . . . . SSU72 . 1 . 3 点号的地方是0,因为0太多了,存成稀疏矩阵可以省点空间。
3.质控
这里是对细胞进行的质控,指标是:
线粒体基因含量不能过高;
nFeature_RNA 不能过高或过低
为什么? nFeature_RNA是每个细胞中检测到的基因数量。nCount_RNA是细胞内检测到的分子总数。nFeature_RNA过低,表示该细胞可能已死/将死或是空液滴。太高的nCount_RNA和/或nFeature_RNA表明“细胞”实际上可以是两个或多个细胞。结合线粒体基因count数除去异常值,即可除去大多数双峰/死细胞/空液滴,因此它们过滤是常见的预处理步骤。 参考自:https://www.biostars.org/p//
3.1 质控指标的可视化
讯享网pbmc[["percent.mt"]] <- PercentageFeatureSet(pbmc, pattern = "^MT-") head(, 3)
orig.ident nCount_RNA nFeature_RNA percent.mt AAACATACAACCAC-1 pbmc3k 2419 779 3.0 AAACATTGAGCTAC-1 pbmc3k 4903 1352 3. AAACATTGATCAGC-1 pbmc3k 3147 1129 0. 讯享网VlnPlot(pbmc, features = c("nFeature_RNA", "nCount_RNA", "percent.mt"), ncol = 3)
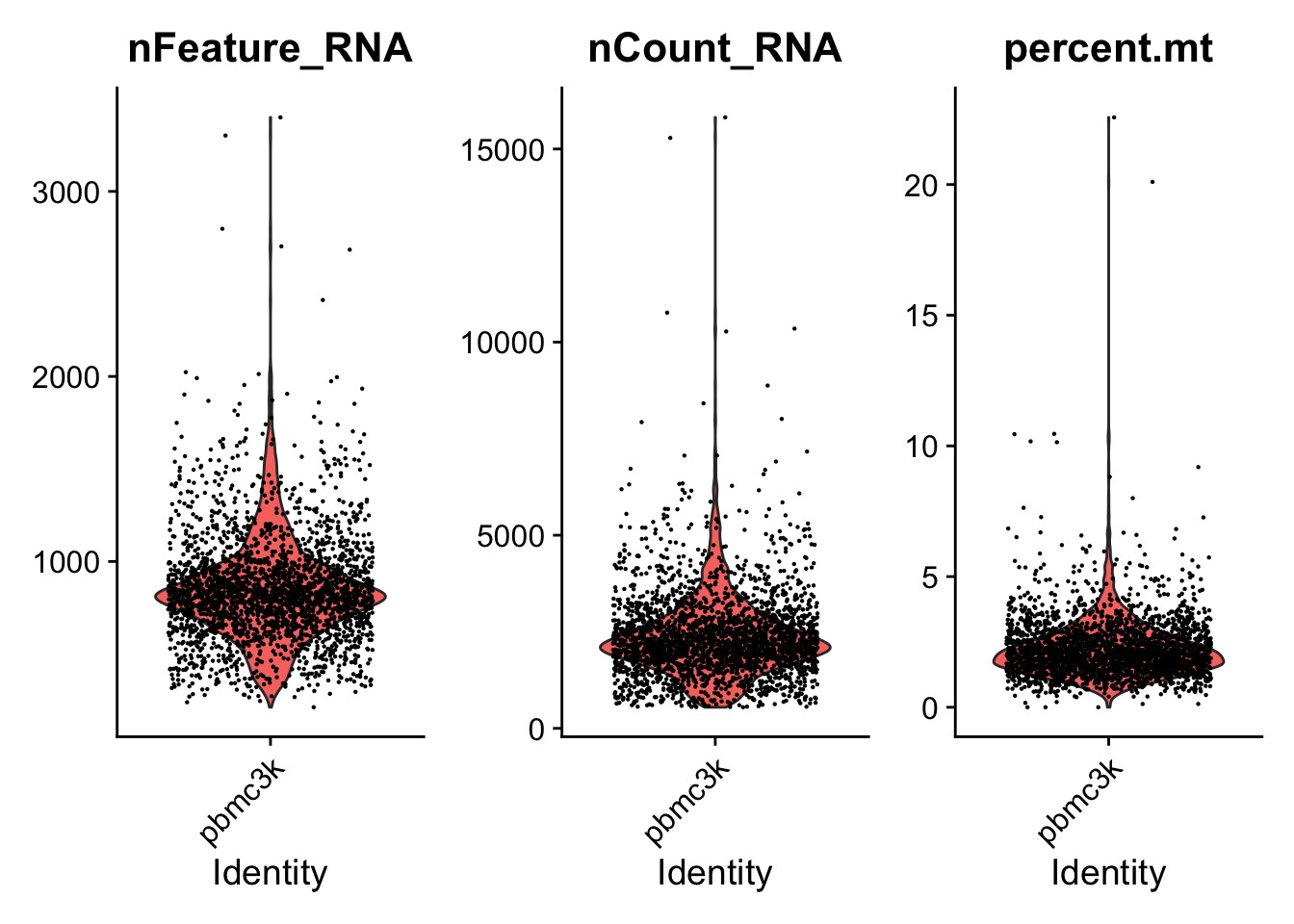
根据这个三个图,确定了这个数据的过滤标准:
nFeature_RNA在200~2500之间;线粒体基因占比在5%以下。
3.2 三个指标之间的相关性
plot1 <- FeatureScatter(pbmc, feature1 = "nCount_RNA", feature2 = "percent.mt") plot2 <- FeatureScatter(pbmc, feature1 = "nCount_RNA", feature2 = "nFeature_RNA") plot1 + plot2 
3.3 过滤
讯享网pbmc <- subset(pbmc, subset = nFeature_RNA > 200 & nFeature_RNA < 2500 & percent.mt < 5) dim(pbmc)
[1] 13714 2638 原本是2700个细胞,可见用上面的标准是过滤掉了62个。
4. 数据归一化,找高变化基因
这里有点类似于常规转录组和芯片数据的差异分析过程,但并不是预知分组的比较,也没有具体的统计量来衡量,直接是你指定用哪种算法,要多少个高变化基因(HVG)
讯享网pbmc <- NormalizeData(pbmc) pbmc@assays$RNA[30:34,1:3]
5 x 3 sparse Matrix of class "dgCMatrix" AAACATACAACCAC-1 AAACATTGAGCTAC-1 AAACATTGATCAGC-1 MRPL20 1. . 1. ATAD3C . . . ATAD3B . . . ATAD3A . . . SSU72 . 1. . 讯享网# 有三种算法:vst、mean.var.plot、dispersion;默认选择2000个HVG pbmc <- FindVariableFeatures(pbmc, selection.method = "vst", nfeatures = 2000) top10 <- head(VariableFeatures(pbmc), 10);top10
[1] "PPBP" "LYZ" "S100A9" "IGLL5" "GNLY" "FTL" "PF4" "FTH1" [9] "GNG11" "S100A8" 这里选了2000个,把前十个在图上标记出来。

讯享网plot1 <- VariableFeaturePlot(pbmc) plot2 <- LabelPoints(plot = plot1, points = top10, repel = TRUE)
When using repel, set xnudge and ynudge to 0 for optimal results 讯享网plot1 + plot2
Warning: Transformation introduced infinite values in continuous x-axis Warning: Transformation introduced infinite values in continuous x-axis 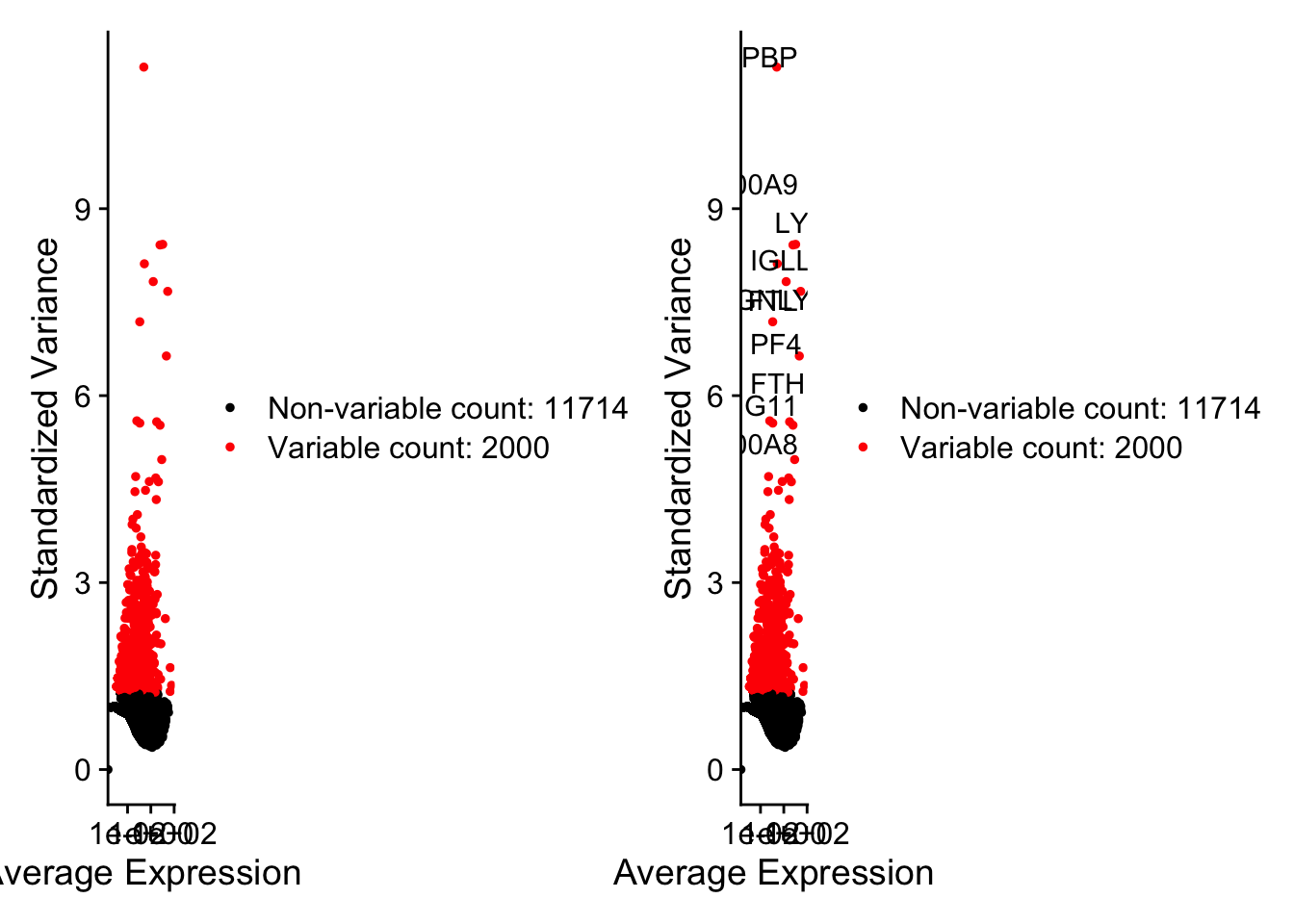
5. 标准化和降维
讯享网all.genes <- rownames(pbmc) pbmc <- ScaleData(pbmc, features = all.genes)
Centering and scaling data matrix 讯享网pbmc[["RNA"]]@scale.data[30:34,1:3]
曾经他们很多都是0。经过处理变成了不同的数字,这个并不会影响到后续分析,因为重点不是绝对的数字,而是比较起来的相对大小。为了让细胞与细胞之间的基因表达量更加可比,做这些处理是合理并可用的。
AAACATACAACCAC-1 AAACATTGAGCTAC-1 AAACATTGATCAGC-1 MRPL20 1. -0. 1. ATAD3C -0.0 -0.0 -0.0 ATAD3B -0. -0. -0. ATAD3A -0. -0. -0. SSU72 -0. 0. -0. ScaleData函数: 调整每个基因的表达量,使整个细胞的平均表达量为0 缩放每个基因的表达,从而使细胞之间的方差为1 此步骤给下游分析中的每个基因相等的权重,因此高表达的基因不会影响全局。
5.1 线性降维PCA
讯享网pbmc <- RunPCA(pbmc, features = VariableFeatures(object = pbmc))
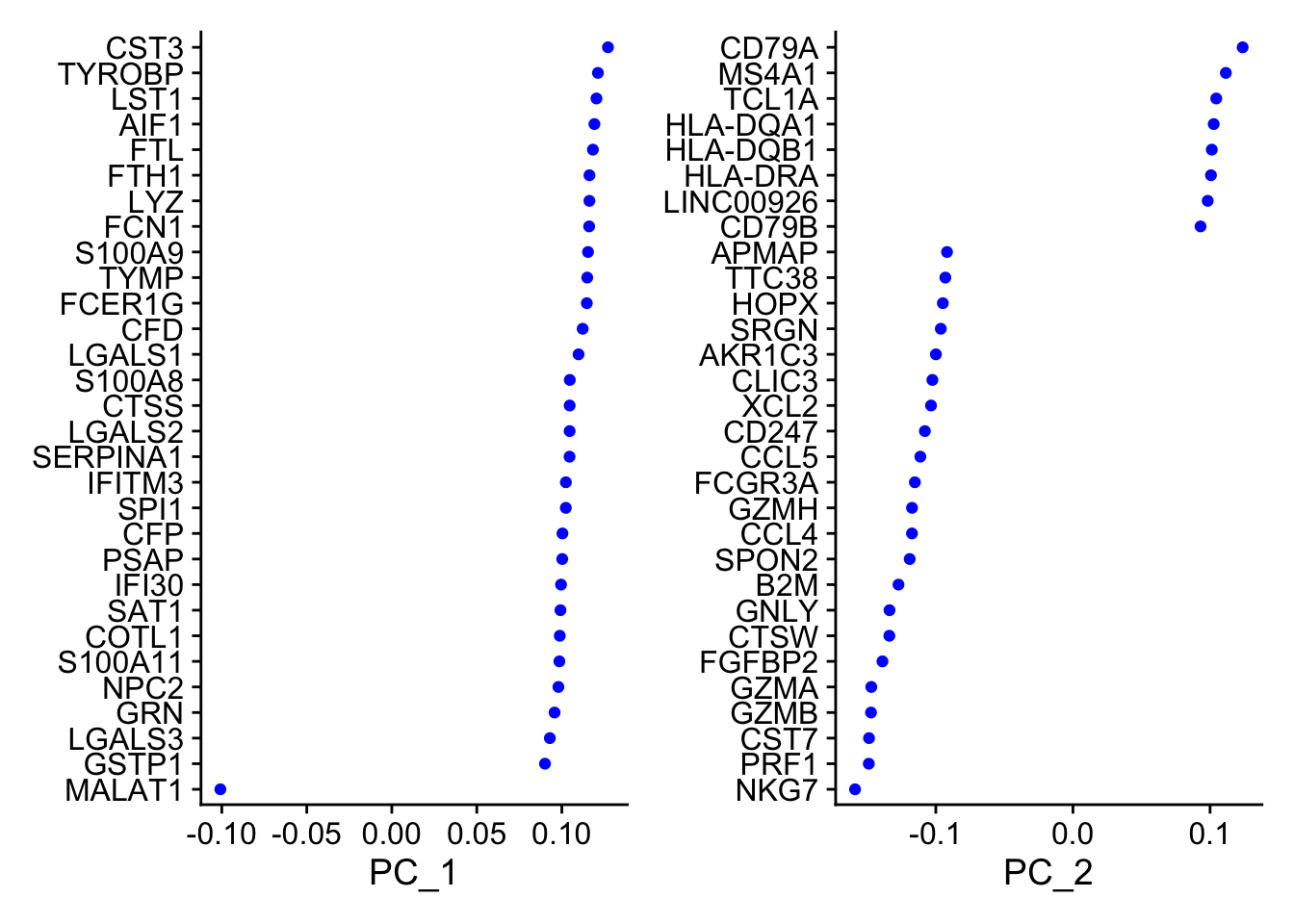
PC_ 1 Positive: CST3, TYROBP, LST1, AIF1, FTL, FTH1, LYZ, FCN1, S100A9, TYMP FCER1G, CFD, LGALS1, S100A8, CTSS, LGALS2, SERPINA1, IFITM3, SPI1, CFP PSAP, IFI30, SAT1, COTL1, S100A11, NPC2, GRN, LGALS3, GSTP1, PYCARD Negative: MALAT1, LTB, IL32, IL7R, CD2, B2M, ACAP1, CD27, STK17A, CTSW CD247, GIMAP5, AQP3, CCL5, SELL, TRAF3IP3, GZMA, MAL, CST7, ITM2A MYC, GIMAP7, HOPX, BEX2, LDLRAP1, GZMK, ETS1, ZAP70, TNFAIP8, RIC3 PC_ 2 Positive: CD79A, MS4A1, TCL1A, HLA-DQA1, HLA-DQB1, HLA-DRA, LINC00926, CD79B, HLA-DRB1, CD74 HLA-DMA, HLA-DPB1, HLA-DQA2, CD37, HLA-DRB5, HLA-DMB, HLA-DPA1, FCRLA, HVCN1, LTB BLNK, P2RX5, IGLL5, IRF8, SWAP70, ARHGAP24, FCGR2B, SMIM14, PPP1R14A, C16orf74 Negative: NKG7, PRF1, CST7, GZMB, GZMA, FGFBP2, CTSW, GNLY, B2M, SPON2 CCL4, GZMH, FCGR3A, CCL5, CD247, XCL2, CLIC3, AKR1C3, SRGN, HOPX TTC38, APMAP, CTSC, S100A4, IGFBP7, ANXA1, ID2, IL32, XCL1, RHOC PC_ 3 Positive: HLA-DQA1, CD79A, CD79B, HLA-DQB1, HLA-DPB1, HLA-DPA1, CD74, MS4A1, HLA-DRB1, HLA-DRA HLA-DRB5, HLA-DQA2, TCL1A, LINC00926, HLA-DMB, HLA-DMA, CD37, HVCN1, FCRLA, IRF8 PLAC8, BLNK, MALAT1, SMIM14, PLD4, LAT2, IGLL5, P2RX5, SWAP70, FCGR2B Negative: PPBP, PF4, SDPR, SPARC, GNG11, NRGN, GP9, RGS18, TUBB1, CLU HIST1H2AC, AP001189.4, ITGA2B, CD9, TMEM40, PTCRA, CA2, ACRBP, MMD, TREML1 NGFRAP1, F13A1, SEPT5, RUFY1, TSC22D1, MPP1, CMTM5, RP11-367G6.3, MYL9, GP1BA PC_ 4 Positive: HLA-DQA1, CD79B, CD79A, MS4A1, HLA-DQB1, CD74, HLA-DPB1, HIST1H2AC, PF4, TCL1A SDPR, HLA-DPA1, HLA-DRB1, HLA-DQA2, HLA-DRA, PPBP, LINC00926, GNG11, HLA-DRB5, SPARC GP9, AP001189.4, CA2, PTCRA, CD9, NRGN, RGS18, GZMB, CLU, TUBB1 Negative: VIM, IL7R, S100A6, IL32, S100A8, S100A4, GIMAP7, S100A10, S100A9, MAL AQP3, CD2, CD14, FYB, LGALS2, GIMAP4, ANXA1, CD27, FCN1, RBP7 LYZ, S100A11, GIMAP5, MS4A6A, S100A12, FOLR3, TRABD2A, AIF1, IL8, IFI6 PC_ 5 Positive: GZMB, NKG7, S100A8, FGFBP2, GNLY, CCL4, CST7, PRF1, GZMA, SPON2 GZMH, S100A9, LGALS2, CCL3, CTSW, XCL2, CD14, CLIC3, S100A12, CCL5 RBP7, MS4A6A, GSTP1, FOLR3, IGFBP7, TYROBP, TTC38, AKR1C3, XCL1, HOPX Negative: LTB, IL7R, CKB, VIM, MS4A7, AQP3, CYTIP, RP11-290F20.3, SIGLEC10, HMOX1 PTGES3, LILRB2, MAL, CD27, HN1, CD2, GDI2, ANXA5, CORO1B, TUBA1B FAM110A, ATP1A1, TRADD, PPA1, CCDC109B, ABRACL, CTD-2006K23.1, WARS, VMO1, FYB 讯享网# 查看前三个主成分由哪些feature组成 print(pbmc[["pca"]], dims = 1:5, nfeatures = 5)
PC_ 1 Positive: CST3, TYROBP, LST1, AIF1, FTL Negative: MALAT1, LTB, IL32, IL7R, CD2 PC_ 2 Positive: CD79A, MS4A1, TCL1A, HLA-DQA1, HLA-DQB1 Negative: NKG7, PRF1, CST7, GZMB, GZMA PC_ 3 Positive: HLA-DQA1, CD79A, CD79B, HLA-DQB1, HLA-DPB1 Negative: PPBP, PF4, SDPR, SPARC, GNG11 PC_ 4 Positive: HLA-DQA1, CD79B, CD79A, MS4A1, HLA-DQB1 Negative: VIM, IL7R, S100A6, IL32, S100A8 PC_ 5 Positive: GZMB, NKG7, S100A8, FGFBP2, GNLY Negative: LTB, IL7R, CKB, VIM, MS4A7 讯享网VizDimLoadings(pbmc, dims = 1:2, reduction = "pca")
#每个主成分对应基因的热图 DimHeatmap(pbmc, dims = 1:15, cells = 500, balanced = TRUE) 
讯享网# 应该选多少个主成分进行后续分析 ElbowPlot(pbmc)
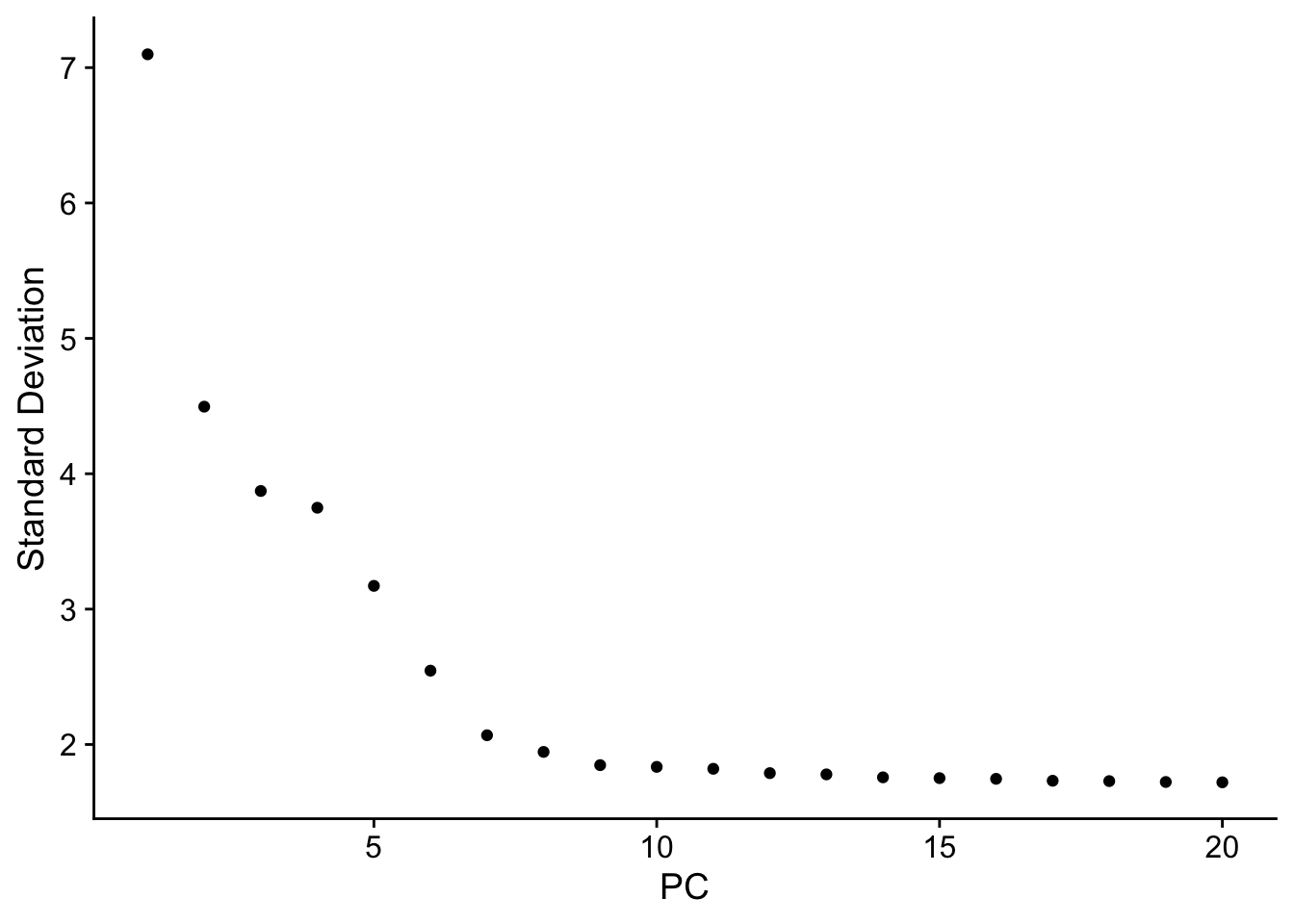
引用一下豆豆:它是根据每个主成分对总体变异水平的贡献百分比排序得到的图,我们主要关注”肘部“的PC,它是一个转折点(也即是这里的PC9-10),说明取前10个主成分可以比较好地代表总体变化
#PC1和2 DimPlot(pbmc, reduction = "pca")+ NoLegend() 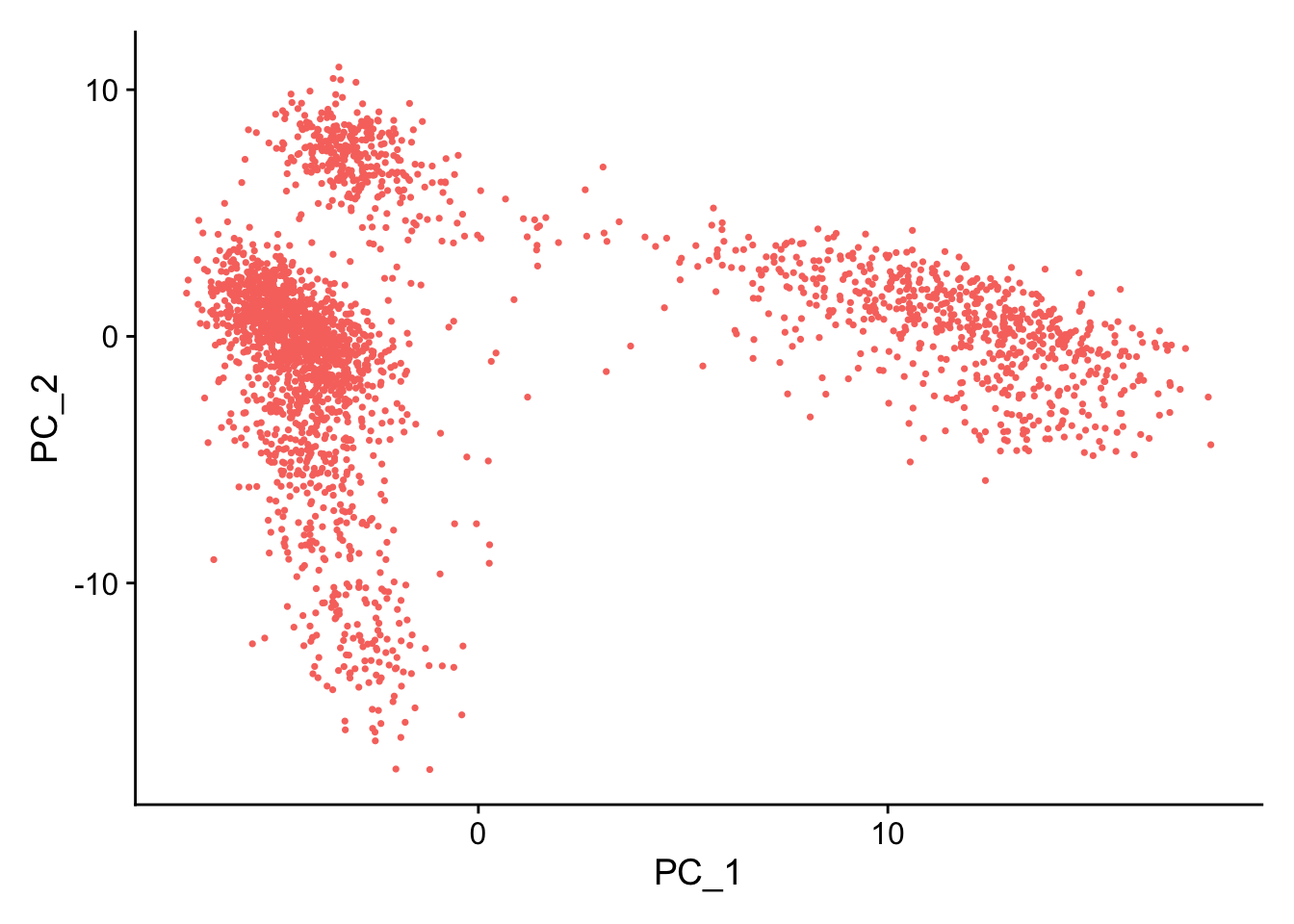
讯享网# 因为我们前面挑选了10个PCs,所以这里dims定义为10个 pbmc <- FindNeighbors(pbmc, dims = 1:10)
Computing nearest neighbor graph 讯享网 Computing SNN
pbmc <- FindClusters(pbmc, resolution = 0.5) #分辨率 讯享网 Modularity Optimizer version 1.3.0 by Ludo Waltman and Nees Jan van Eck Number of nodes: 2638 Number of edges: 95965 Running Louvain algorithm... Maximum modularity in 10 random starts: 0.8723 Number of communities: 9 Elapsed time: 0 seconds
# 结果聚成几类,用Idents查看 length(levels(Idents(pbmc))) 讯享网 [1] 9
resolution的大小决定着分群的数量,3000细胞时,0.4~1.2的分辨率表现较好。
5.2 UMAP 和t-sne
PCA是线性降维,这两个是非线性降维。理解起来比较复杂,用起来只是一个函数。
pbmc <- RunUMAP(pbmc, dims = 1:10) 讯享网 Warning: The default method for RunUMAP has changed from calling Python UMAP via reticulate to the R-native UWOT using the cosine metric To use Python UMAP via reticulate, set umap.method to 'umap-learn' and metric to 'correlation' This message will be shown once per session
14:28:12 UMAP embedding parameters a = 0.9922 b = 1.112 讯享网 14:28:12 Read 2638 rows and found 10 numeric columns
14:28:12 Using Annoy for neighbor search, n_neighbors = 30 讯享网 14:28:12 Building Annoy index with metric = cosine, n_trees = 50
0% 10 20 30 40 50 60 70 80 90 100% 讯享网 [----|----|----|----|----|----|----|----|----|----|
| 14:28:12 Writing NN index file to temp file /var/folders/mg/s3ln5v717ps4934ym_3zk80000gn/T//RtmpWoykmm/filef4d08 14:28:12 Searching Annoy index using 1 thread, search_k = 3000 14:28:13 Annoy recall = 100% 14:28:13 Commencing smooth kNN distance calibration using 1 thread 14:28:13 Initializing from normalized Laplacian + noise 14:28:13 Commencing optimization for 500 epochs, with positive edges 14:28:16 Optimization finished 讯享网DimPlot(pbmc, reduction = "umap")
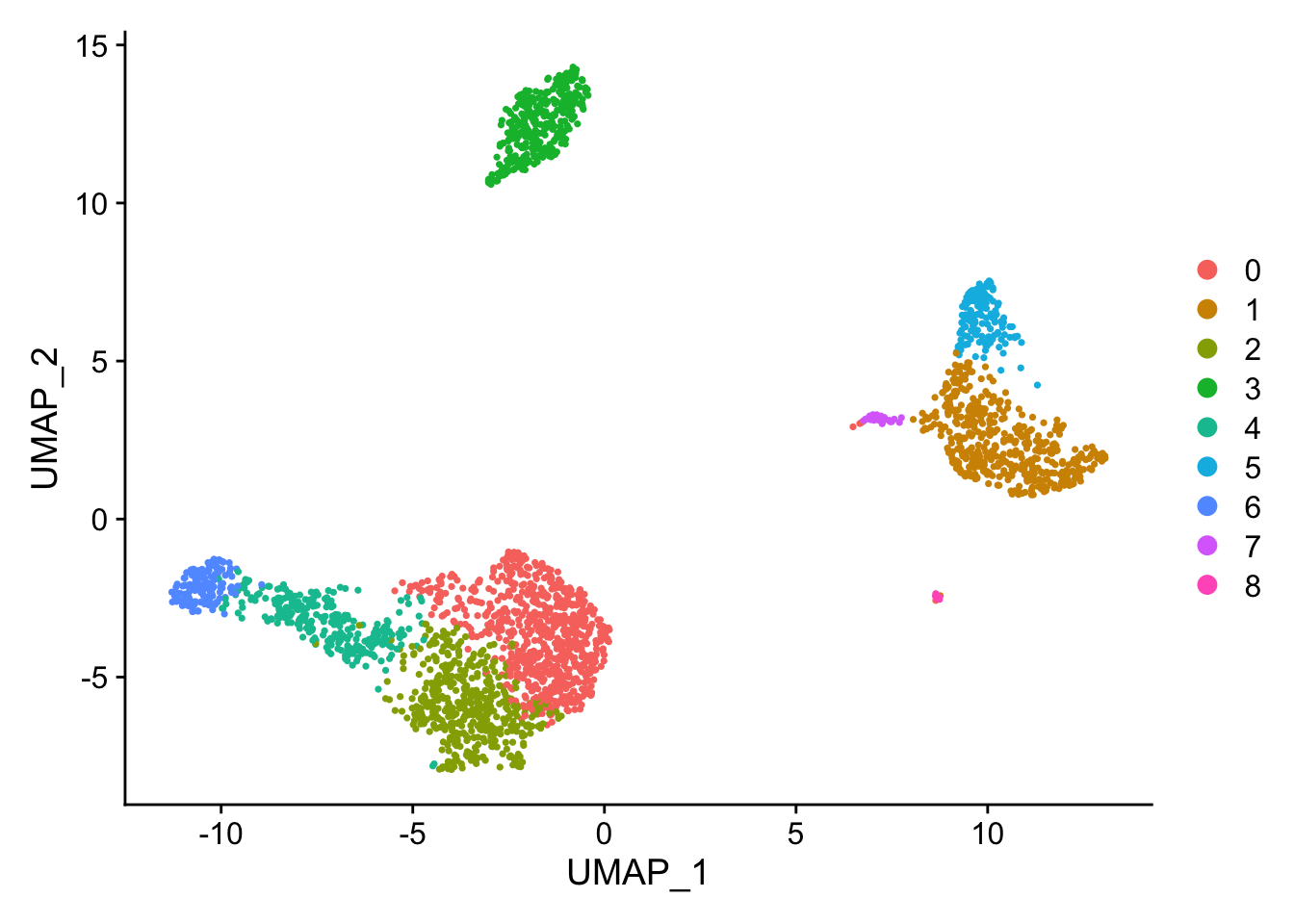
saveRDS(pbmc, file = "01_data/pbmc_tutorial.rds") 6.找marker基因
啥叫marker基因呢。和差异基因里面的上调基因有点类似,某个基因在某一簇细胞里表达量都很高,在其他簇表达量很低,那么这个基因就是这簇细胞的象征。
现成的函数可以实现:
- 单独找某个cluster的maker基因
- 比较某几个cluster
- 找全部cluster的maker基因
讯享网cluster1.markers <- FindMarkers(pbmc, ident.1 = 1, min.pct = 0.25) head(cluster1.markers, n = 3)
p_val avg_log2FC pct.1 pct.2 p_val_adj S100A9 0 5. 0.996 0.215 0 S100A8 0 5. 0.975 0.121 0 FCN1 0 3. 0.952 0.151 0 讯享网cluster5.markers <- FindMarkers(pbmc, ident.1 = 5, ident.2 = c(0, 3), min.pct = 0.25) head(cluster5.markers, n = 3)
p_val avg_log2FC pct.1 pct.2 p_val_adj FCGR3A 2.e-209 4. 0.975 0.039 2.e-205 IFITM3 6.e-199 3. 0.975 0.048 8.e-195 CFD 8.e-198 3. 0.938 0.037 1.e-193 讯享网pbmc.markers <- FindAllMarkers(pbmc, only.pos = TRUE, min.pct = 0.25, logfc.threshold = 0.25) pbmc.markers %>% group_by(cluster) %>% top_n(n = 2, wt = avg_log2FC)
Registered S3 method overwritten by 'cli': method from print.boxx spatstat.geom 讯享网 # A tibble: 18 x 7 # Groups: cluster [9] p_val avg_log2FC pct.1 pct.2 p_val_adj cluster gene <dbl> <dbl> <dbl> <dbl> <dbl> <fct> <chr> 1 1.74e-109 1.07 0.897 0.593 2.39e-105 0 LDHB 2 1.17e- 83 1.33 0.435 0.108 1.60e- 79 0 CCR7 3 0\. 5.57 0.996 0.215 0\. 1 S100A9 4 0\. 5.48 0.975 0.121 0\. 1 S100A8 5 7.99e- 87 1.28 0.981 0.644 1.10e- 82 2 LTB 6 2.61e- 59 1.24 0.424 0.111 3.58e- 55 2 AQP3 7 0\. 4.31 0.936 0.041 0\. 3 CD79A 8 9.48e-271 3.59 0.622 0.022 1.30e-266 3 TCL1A 9 1.17e-178 2.97 0.957 0.241 1.60e-174 4 CCL5 10 4.93e-169 3.01 0.595 0.056 6.76e-165 4 GZMK 11 3.51e-184 3.31 0.975 0.134 4.82e-180 5 FCGR3A 12 2.03e-125 3.09 1 0.315 2.78e-121 5 LST1 13 1.05e-265 4.89 0.986 0.071 1.44e-261 6 GZMB 14 6.82e-175 4.92 0.958 0.135 9.36e-171 6 GNLY 15 1.48e-220 3.87 0.812 0.011 2.03e-216 7 FCER1A 16 1.67e- 21 2.87 1 0.513 2.28e- 17 7 HLA-DPB1 17 7.73e-200 7.24 1 0.01 1.06e-195 8 PF4 18 3.68e-110 8.58 1 0.024 5.05e-106 8 PPBP
6.1 比较某个基因在几个cluster之间的表达量
小提琴图
VlnPlot(pbmc, features = c("MS4A1", "CD79A")) 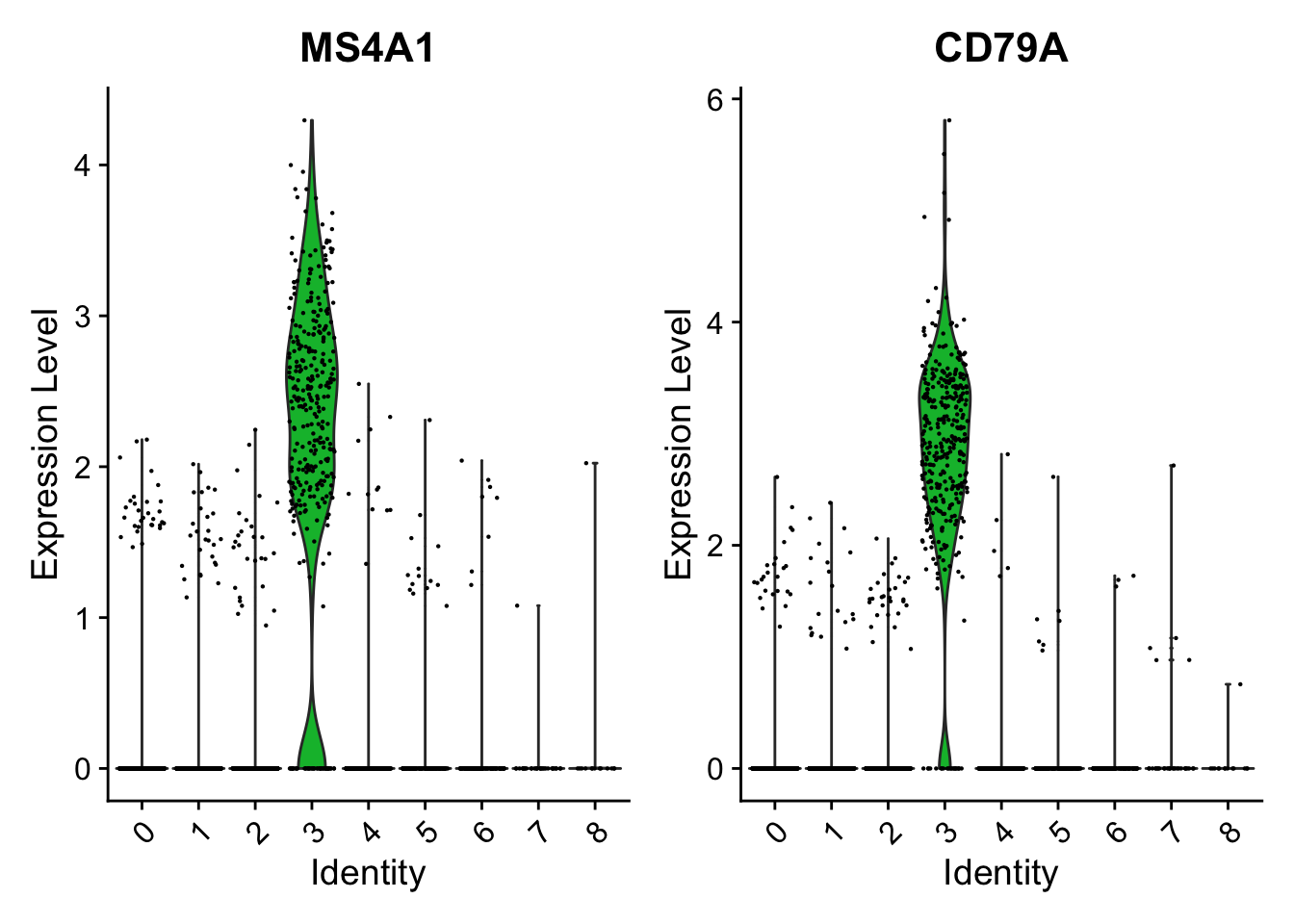
讯享网#可以拿count数据画 VlnPlot(pbmc, features = c("NKG7", "PF4"), slot = "counts", log = TRUE)
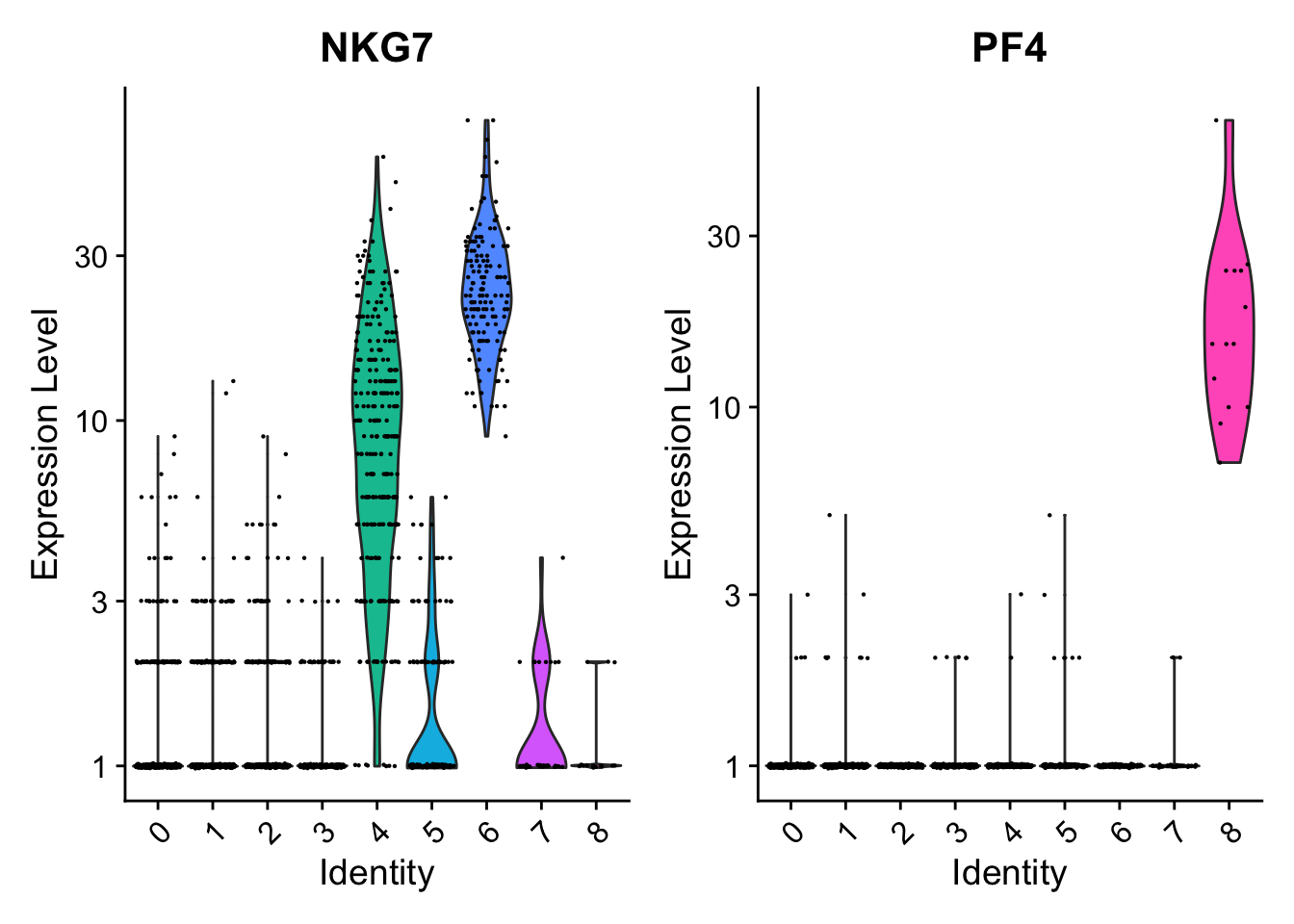
在umap图上标记
FeaturePlot(pbmc, features = c("MS4A1", "GNLY", "CD3E", "CD14", "FCER1A", "FCGR3A", "LYZ", "PPBP", "CD8A")) 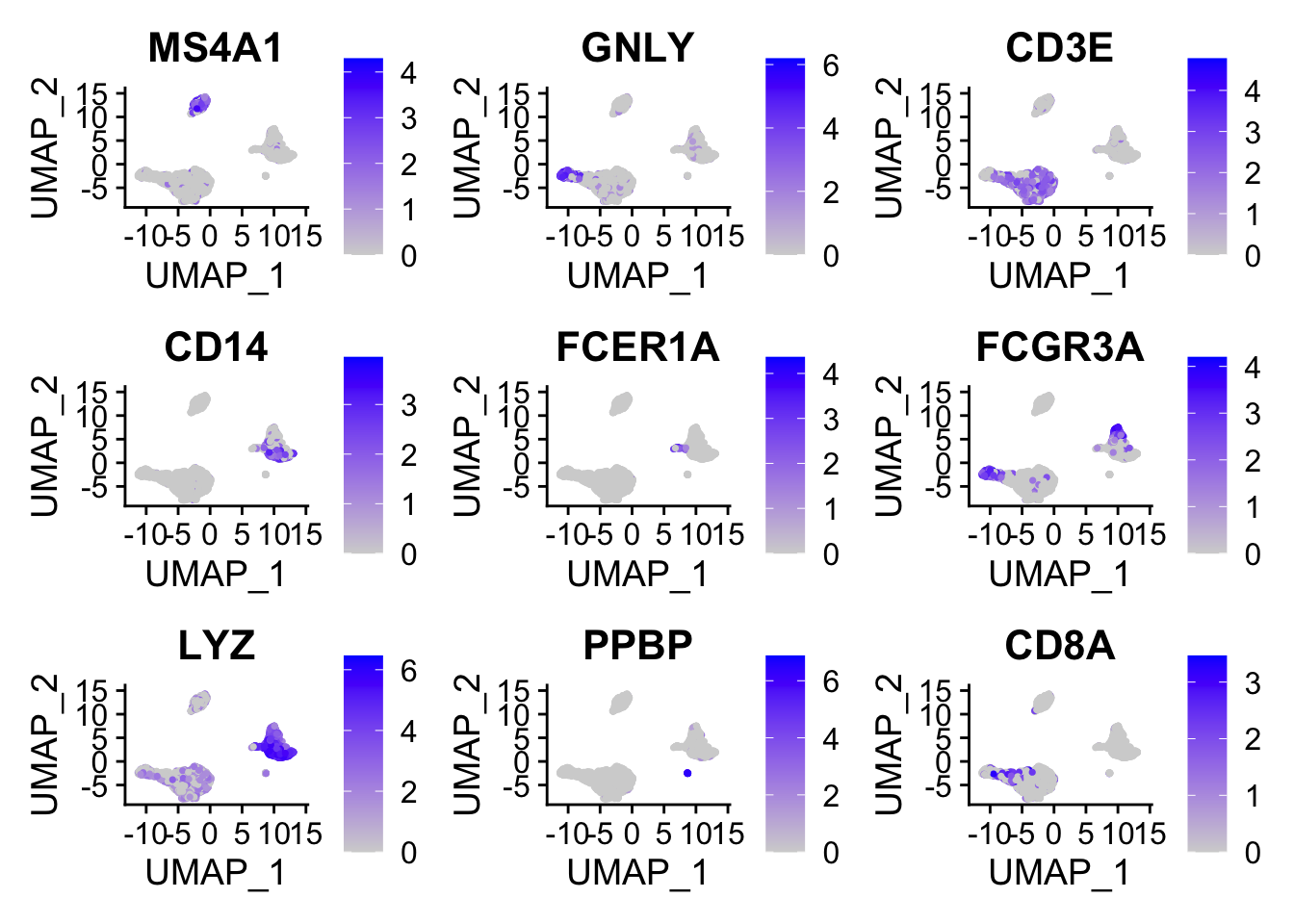
讯享网top10 <- pbmc.markers %>% group_by(cluster) %>% top_n(n = 10, wt = avg_log2FC)
6.2 marker基因的热图
DoHeatmap(pbmc, features = top10$gene) + NoLegend() 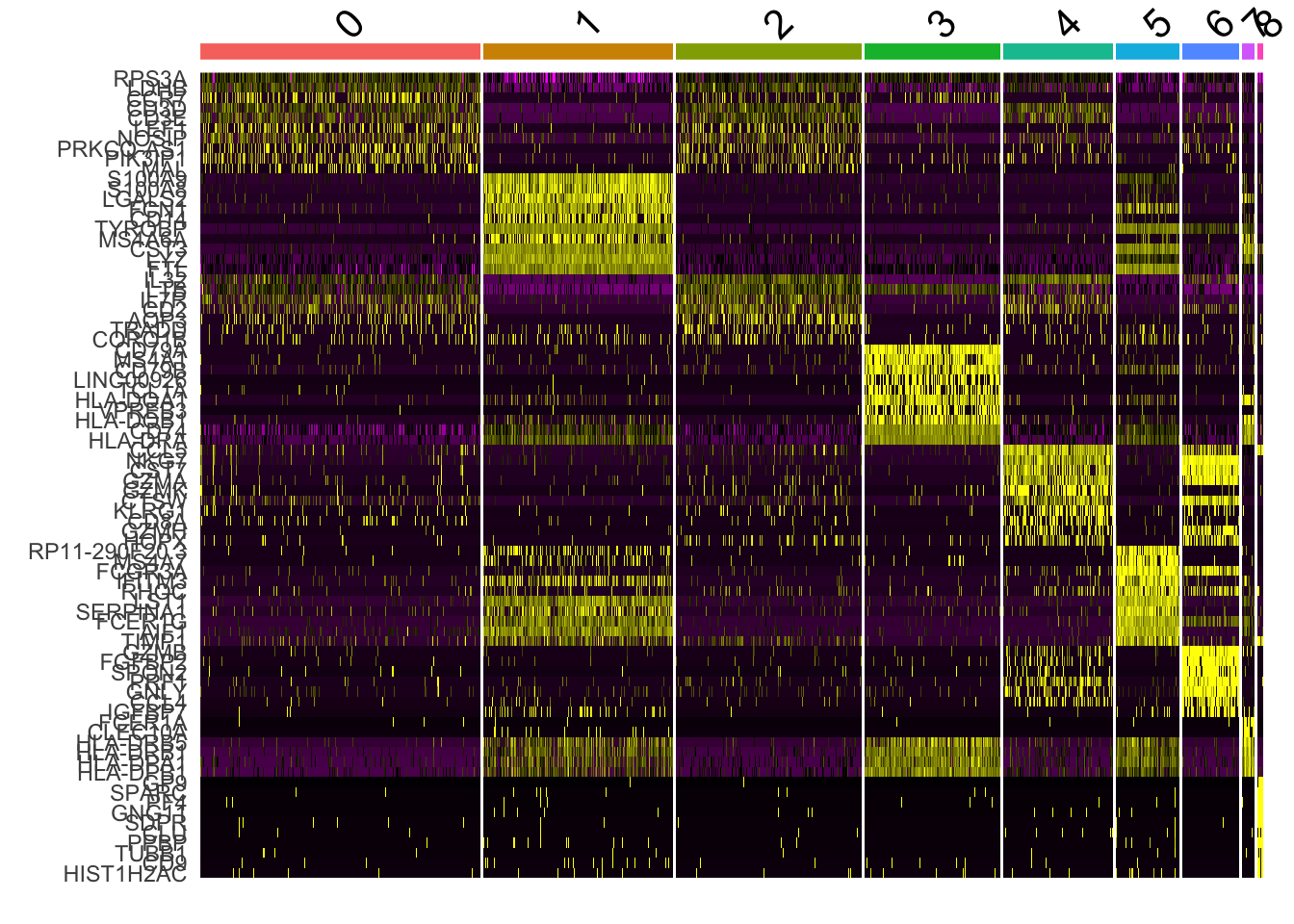
7. 根据marker基因确定细胞
在示例数据里,这一步找到哪些基因对应什么细胞的表格,是现成的。我问了豆豆,上哪里找这样的对应信息呢?豆豆说有专门的R包–singleR来做细胞注释,并且需要根据这些注释多次调整前面的参数,使聚类的结果与生物学意义对接上。标准操作会R语言就能搞定,生物学意义还需要慢慢磨啊。整理完三大R包,我就研究singleR。
讯享网new.cluster.ids <- c("Naive CD4 T", "CD14+ Mono", "Memory CD4 T", "B", "CD8 T", "FCGR3A+ Mono", "NK", "DC", "Platelet") names(new.cluster.ids) <- levels(pbmc) pbmc <- RenameIdents(pbmc, new.cluster.ids) DimPlot(pbmc, reduction = "umap", label = TRUE, pt.size = 0.5) + NoLegend()
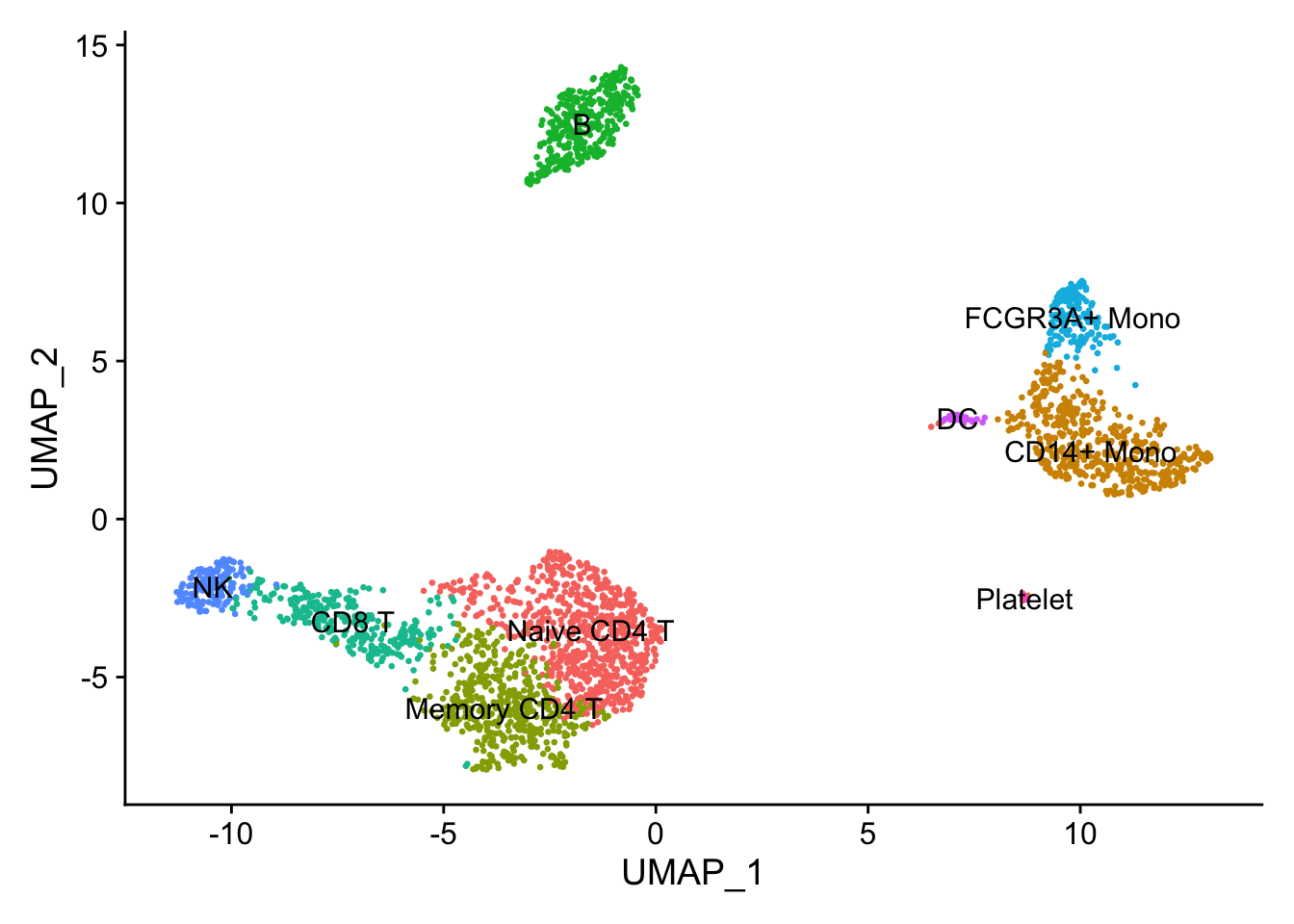
版权声明:本文内容由互联网用户自发贡献,该文观点仅代表作者本人。本站仅提供信息存储空间服务,不拥有所有权,不承担相关法律责任。如发现本站有涉嫌侵权/违法违规的内容,请联系我们,一经查实,本站将立刻删除。
如需转载请保留出处:https://51itzy.com/kjqy/32747.html