
目录
遗传学知识
数量性状
分子标记(Molecular Marker)
DNA分子标记的特点
QTL定位
QTL(quantitative trait locus)
QTL定位的主要流程:
全基因组关联分析
GWAS(Genome-Wide Association Study,全基因组关联分析)
GWAS的研究流程:
曼哈顿图和-plot
知识补充
1.LOD值:Logarithm off the odds score
2.QTL的统计方法:
3.GWAS为什么使用混合线性模型?
4. 哪些因素会影响GWAS和QTL定位的精度?
遗传学知识
-
数量性状
在一个群体内表现为连续变异的性状,相对性状间没有质的差异,如身高、体重、株高等。质量性状的定义与其相反,比如豌豆的皱粒和圆粒、果蝇的红眼和白眼等。此外,还有一种性状属于数量性状,表型呈非连续变异,而遗传物质的数量呈潜在的连续变异的性状,即只有超过某一遗传阈值时才出现的性状,如动植物包括人类的抗病力、死亡率以及单胎动物的产仔数等性状,称为阈性状(threshold character或threshold trait)
表1 质量性状和数量性状对比
| 质量性状 |
数量性状 |
|
| 变异 |
不连续 |
连续 |
| 相对性状 |
有显隐性 |
无显隐性,杂合表现中间值 |
| 环境因素 |
不易受影响 |
对环境变化敏感 |
| 研究方法 |
直接观察、文字记载 |
需要度量,依靠群体,必须使用统计方法 |
| 孟德尔遗传定律 |
符合 |
不符合,但决定性状的每对微效基因的遗传符合 |
-
分子标记(Molecular Marker)
是以个体间遗传物质的核苷酸序列变异为基础的遗传标记。主要包含三类:
(1)以Southern杂交为核心的第一代分子标记--Restriction Fragment Length Polymorphism(RFLP,限制性片段长度多态性)


(2)以PCR扩增为核心的第二代分子标记--Randomly Amplified Polymorphic DNA(RAPD,随机扩增多态性)

(3)以单核苷酸多态性(SNP)为核心的第三代分子标记


表2 各种分子标记优缺点对比
| RFLP(第一代) |
RAPD(第二代) |
SNP(第三代) |
|
| 优点 |
共显性、稳定性好 |
1.对DNA的质量和数量要求 2.引物具有广泛性和通用性 3.快速和经济 4.无需基因组任何分子信息 5.试验材料无需有性世代,可用任何单一亲本 |
1.SNP数量多,分布广泛 2.稳定性高 3.共显性
4.适于快速、规模化筛查 |
| 缺点 |
1.DNA纯度要求高 2.用量大 3.步骤繁琐 4.探针制备和保存不方便 5.同位素标记有放射性 |
1.显性:无法区分纯合体和杂合体 2.扩增产物的稳定性和重复性差 |
如果用测序或DNA芯片杂交的方法,成本较高 |

共显性是指杂合F1代个体中,两个纯合亲本的等位基因都能表现出来的标记,即能够区分纯合和杂合的标记;而显性标记只能检测显性等位基因。
-
DNA分子标记的特点
DNA分子标记能直接反映生物个体或种群间基因组中某种差异特征的DNA片段,特点有:
(1)不受组织、发育阶段、季节和环境的影响,与表达与否无关;
(2)数量多,遍布整个基因组,涉及基因座位多
(3)自然存在许多等位变异,多态性高
(4)表现中性,补一下目标基因表达
(5)共显性--区分纯合体和杂合体
QTL定位
-
QTL(quantitative trait locus)
即数量性状位点,代表染色体上影响数量性状的某个区段,区段内可能会有一个甚至多个影响数量性状的功能基因。QTL定位指检查分子标记与QTL间的连锁关系,并估算QTL的表型效应。其本质是利用功能基因与分子标记间的连锁和重组,实现对功能基因位置的定位。近几年QTL定位应用的较为广泛,在人类基因上与疾病有关的基因位点甚多;植物上,模式植物抗逆性基因的定位较多。国内在家畜基因组学上的QTL专家有中国农业大学的张勤教授、华中农业大学的熊远著院士。
-
QTL定位的主要流程:
(1)构建分离群体(作图群体):注意亲本的选择,要有稳定的表型差异;注意群体的大小、分离群体的类型,是否有表型重复性,考虑QTL的显隐性
(2)遗传标记检测和表型测定(需要准确的表型数据)
(3)利用基因型图谱或分子标记和表型数据进行连锁分析:通过观察LOD曲线的峰值,进行QTL的初步定位
(4)QTL的精细定位,使用统计分析鉴定候选基因
全基因组关联分析
-
GWAS(Genome-Wide Association Study,全基因组关联分析)
GWAS是连接遗传变异和表型的主要工具。它的理论基础是连锁不平衡(Linkage Disequilibrium,LD)。LD是指给定种群中不同基因座(位点)上的等位基因之间的非随机关联性,即分属两个或两个以上基因座位的等位基因同时出现的频率高于预期的现象。可用TASSEL进行GWAS分析。
-
GWAS的研究流程:
(1)自然群体资源收集和鉴定(纯化)
(2)获取目标性状的表型数据
(3)群体重测序(全基因组、转录组、外显子组、GBS等),获取基因型数据(测序读段比对、遗传变异如SNP的识别、GWAS变异标记(如SNP)构建)
(4)基因型补缺和过滤,表型数据分析
(5)遗传多样性、群体结构、亲缘关系、LD等分析
群体结构是由于个体间非随机交配导致的群体亚群之间等为基因频率的系统差异。这种系统差异是GWAS 中影响很大的混淆变量,可造成很大的假阳性。
LD是指Linkage Disequilibrium,指给定种群中不同基因座(位点)上的等位基因之间的非随机关联性,即分属两个或两个以上基因座位的等位基因同时出现的概率高于预期的现象。与连锁的区别是:1)连锁指的是位于同一条染色体上的基因连锁遗传,LD是指群体内等位基因之间的相关,即连锁是“个体”上两位点物理距离接近,LD 是“群体”上相关。2)紧密连锁可以导致 LD,还有自然选择、遗传漂变、群体结构等导致 LD。
(6)关联分析
(7)候选基因筛选与挖掘
(8)实验验证、分子机制解析
-
曼哈顿图和-plot
使用GWAS分析软件如TASSEL可以生成曼哈顿图(Manhattan)或-plot图来评估模型的准确性和发现潜在SNP位点。
比如下面两张图是针对EarHT表型的分析图:
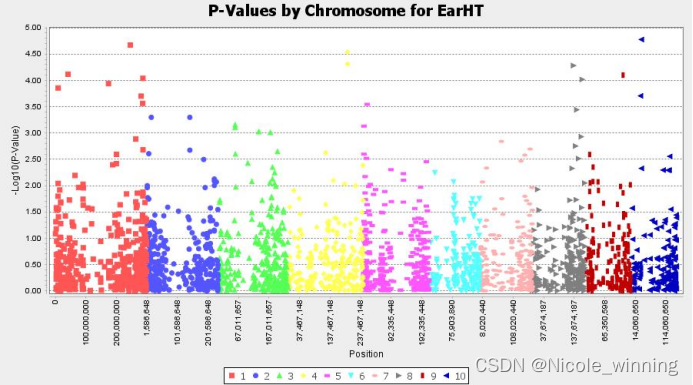
上图为曼哈顿图,曼哈顿图用于可视化基因组中每个位点的关联统计显著性。其中X轴表示基因组的位置,y轴代表统计学的显著性(以-log10(p)表示,p-value越小,则值越大)。图中每个点代表基因组中的一个位点,点的高度表示该位点的关联显著性,点越高说明该基因区域(该染色体)与表型或疾病关联程度更强。从曼哈顿图中可以看出,1、4、8、10号染色体与性状EarHT的关联性更强。

上图为-plot图,-plot指Quantile-Quantile Plot,可以用于直观验证一组或两组数据是否来自同一分布。上面的-plot中,X轴表示期望-Log10(p-value)(值越大,说明p-value越小,即越显著),Y轴表示观测的-Log10(p-value),由上图可以看出,在p-value较小时,观测p值和期望p值比较一致,-plot的左下角是显著性低的点,它们与EarHT性状不关联,与45°对角线拟合说明模型合理;而当期望p-value增大时,发现点慢慢偏离对角线,且位于对角线上方,说明观测到的p-value远比期望的更小,即更显著。即-plot的右上角是显著性高的位点,且观测p-value相对期望p-value小得多,说明是潜在的与EarHT性状相关的位点。
知识补充
1.LOD值:Logarithm off the odds score
LOD = log10(L1/L0),L1是该位点有QTL的概率,L0是该位点无QTL的概率。所以LOD值越大,说明该位点有QTL的概率大于无QTL的概率,即该位点是候选基因位点。
比如下图是针对玉米MAGIC群体的株高(Plant Height,PH)表型的LOD曲线,可以看到峰值在第6、8、10号染色体,说明这三个染色体可能与PH表型有关。

那么LOD值多少为大呢,即LOD阈值设为多少可以认为该位点有QTL呢?这就需要使用Permutation test置换检验来确定显著性阈值。Permutation test与Bootstrap的区别是,前者为不放回抽样,后者为放回抽样。在这里Permutation test的方法是,先随机打乱表型与基因型之间的对应关系,接着进行QTL分析记录区间中最大的LOD值,重复该过程1000次,得到“表型-基因型”不关联的情况下,LOD值的分布情况;取随机模型得到的前5%或1%为显著性阈值。(比如下图的置换检验得到LOD阈值为3左右)


2.QTL的统计方法:
1)单标记分析:方差分析(检验两个及两个以上样本均数差别的显著性)、t检验、线性回归
2)区间作图法(IM):在线性模型基础上,利用最大似然法对相邻标记构成的区间内任意一点可能存在的QTL进行似然比检验,进而获得其效应的极大似然估计。
3)复合区间作图法(CIM):使用逐步回归,将其它与表型相关的QTL作为协变量控制背景遗传效应。
3.GWAS为什么使用混合线性模型?
混合线性模型是既包含固定效应,又包含随机效应的线性模型,与简单线性模型相比,增加了随机效应,用于表征群体内个体间异质的亲缘关系造成的多基因效应,能够修正亲缘关系和群体结构,从而降低群体结构的假阳性。

4. 哪些因素会影响GWAS和QTL定位的精度?
(1)重组事件的数目:重组的越多,定位越精准
(2)分子标记的数目:需要比重组事件数目多
(3)群体的大小:群体大了,重组事件才多
(4)群体的类型:不同群体的重组率差别很大;是否有杂合基因型的影响
(5)群体结构的影响:群体隔离和受选择会降低重组率,且可能会有假阳性
(6)模型是否合适,GWAS 使用混合线性模型,相比线性模型考虑了随机效应,可降低假阳性
(7)位点之间是否有复杂的互作
(8)是否有测量误差
参考资料:
https://zhuanlan.zhihu.com/p/?utm_id=0
https://b23.tv/UTEoaEb
版权声明:本文内容由互联网用户自发贡献,该文观点仅代表作者本人。本站仅提供信息存储空间服务,不拥有所有权,不承担相关法律责任。如发现本站有涉嫌侵权/违法违规的内容,请联系我们,一经查实,本站将立刻删除。
如需转载请保留出处:https://51itzy.com/kjqy/21717.html