
小数据教程-数据分析
使用GBLUP和交叉验证进行预测
使用GFBLUP和交叉验证进行预测
基因集富集分析
一、使用GBLUP和交叉验证进行预测
这里,我们将展示如何使用qgg中的greml函数执行基因组**线性无偏预测(GBLUP)分析和交叉验证。这包括在训练集中使用限制最大似然估计(REML)估计方差成分,在验证集中使用GBLUP进行预测。这将在果蝇遗传参考面板(DGRP)中可用的“抗饥饿”表型和基因型数据中得到说明。
要执行GBLUP分析,以下输入数据是必不可少的。
y: vector of phenotype
X: design matrix for covariables
W: centered and scaled genotype matrix
G: genomic relationship matrix
该脚本包括以下步骤:
1)加载和准备数据
2)用于估计方差成分的限制最大似然(REML)分析
3)使用GBLUP模型的REML分析和交叉验证。
#library(devtools) #install_github("psoerensen/qgg") library(qgg) 讯享网
加载并准备GBLUP分析数据
加载表型和共变量数据
讯享网load(file = "./phenotypes/starv_inv_wo.Rdata") dim(starv) [1] 406 20 head(starv) L sex y In2Lt In2RNS In2RY1 In2RY2 In2RY3 In2RY4 In2RY5 In2RY6 1 100 M 49.28000 INV/ST ST ST ST ST ST ST ST 2 101 M 47.20000 INV/ST ST ST ST ST ST ST ST 3 105 M 51.04000 ST ST ST ST ST ST ST ST 4 109 M 44.96000 INV/ST ST ST ST ST ST ST ST 5 129 M 33.08475 ST ST ST ST ST ST ST ST 6 136 M 63.04000 ST ST ST ST ST ST ST ST In2RY7 In3LP In3LM In3LY In3RP In3RK In3RMo In3RC wo 1 ST ST ST ST ST INV ST ST y 2 ST ST ST ST ST ST ST ST n 3 ST ST ST ST ST INV ST ST n 4 ST ST ST ST ST ST ST ST n 5 ST ST ST ST ST ST ST ST n 6 ST INV/ST ST ST ST INV/ST ST ST y
创建一个抗饥饿表型的向量y。
data <- starv y <- data$y 为共变量准备设计矩阵X。fm是用于在模型中包含相关变量以构建设计矩阵的公式。
讯享网fm <- y ~ wo +In2Lt + In2RNS + In3RP + In3RK + In3RMo X <- model.matrix(fm, data=data) dim(X) [1] 406 12 X[1:5,] (Intercept) woy In2LtINV/ST In2LtST In2RNSINV/ST In2RNSST In3RPINV/ST 1 1 1 1 0 0 1 0 2 1 0 1 0 0 1 0 3 1 0 0 1 0 1 0 4 1 0 1 0 0 1 0 5 1 0 0 1 0 1 0 In3RPST In3RKINV/ST In3RKST In3RMoINV/ST In3RMoST 1 1 0 0 0 1 2 1 0 1 0 1 3 1 0 0 0 1 4 1 0 1 0 1 5 1 0 1 0 1
加载中心和缩放基因型矩阵W
load(file= "./genotypes/dgrp2_W2.Rdata") dim(W) [1] 205 W[1:5,1:5] 2L_5317 2L_5372 2L_5390 2L_5403 2L_5465 21 -0. -0. 1. -0. -0. 26 -0. -0. -0. -0. -0. 28 -0. -0. 1. -0. -0. 31 4.0006648 -0. -0. -0. -0. 32 -0. -0. -0. -0. -0. 加性基因组关系矩阵G(VanRaden PM. 2008. J Dairy Sci. 91:4414-4423) 使用所有遗传标记构建如下:
G=WW′/m 其中W为中心和缩放的基因型矩阵,m为标记总数。
从基因型矩阵W计算累加的基因组关系矩阵G。
讯享网L <- data$L G <- grm(W=W) G <- G[L,L] # 这确保了G中的行id对应于数据中的行id,每一行都有雄性和雌性数据
限制最大似然(REML)分析
greml函数的幕后工作
REML分析用于估计方差分量,σ2e σ2e是线性混合模型中的随机效应:
y=Xb+Zg+e
其中y是表型观察的向量,X和Z为固定和随机效应的设计矩阵,b是固定效应向量,g是所有遗传标记捕获的基因组值的载体,e是残差向量。随机基因组值和残差假设为独立的正态分布值,如下所述:g∼N(0,Gσ2g) and e∼N(0,Iσ2e). 因此,我们假设观察到的表现型 y∼N(Xb,V) ;V=ZGZ′σ2g+Iσ2e .
在此,我们根据在整个研究人群中观察到的表型,使用GBLUP预测遗传价值:
g=σ2g GZ′V−1(y−Xb)
表型预测为:
y=Xb^+Zg^
greml函数在收敛之前要经过多次迭代(即,连续轮之间的参数变化小于指定的阈值,请参阅greml帮助页面中的“tol”参数)。在本例中,每次迭代都返回方差组件(第3列)和(第4列)的值。
greml函数返回一个列表结构,其中包括对固定效果(b)、随机效果(g)和剩余效果(e)的估计。列表中的其他值在greml帮助页面中描述。将verbose = FALSE更改为verbose = TRUE,可以查看迭代。
fitG <- greml(y=y, X=X, GRM=list(G=G), verbose = FALSE) REML使用GBLUP模型进行分析和交叉验证
根据训练和验证群体之间的基因组关系来预测遗传价值:

讯享网
基因组的关系矩阵:

根据训练(t)数据Gtt中的个体之间、验证(v)数据Gvv中的个体之间以及训练和验证数据Gvt中的个体之间的关系进行划分。因此,在训练数据中使用估计的方差成分(檩子2g和檩子2e)来预测总基因组易感性。最右边的术语(yt−Xtbt)构成了训练数据中个体为固定效应而校正的表型。反项V^ - 1t本质上是校正后的表型的方差-协方差结构。这两个术语相乘就是训练数据中个体的标准化和校正的表型,这些表型被投射到训练数据和验证数据之间的总遗传协方差结构上。
估计训练集中观察到的表型的方差成分。根据估计的方差分量,预测验证集的表型为:

通过从1 - 406 (y的长度)中随机取样30个值并重复此取样50次,可以创建50个验证集。验证集保存在验证矩阵中。这个矩阵指定GREML分析中使用的数据行。
讯享网n <- length(y) validate <- replicate(50, sample(1:n, 30)) cvG <- greml(y = y, X = X, GRM = list(G=G), validate = validate) str(cvG) List of 4 $ accuracy:'data.frame': 50 obs. of 7 variables: ..$ Corr : num [1:50] 0.087 -0.049 -0.058 0.388 -0.026 0.264 0.277 -0.004 0.161 0.202 ... ..$ R2 : num [1:50] 0.008 0.002 0.003 0.15 0.001 0.07 0.077 0 0.026 0.041 ... ..$ Nagel R2 : num [1:50] NA NA NA NA NA NA NA NA NA NA ... ..$ AUC : num [1:50] NA NA NA NA NA NA NA NA NA NA ... ..$ intercept: num [1:50] 50.8 52.7 54.5 49.1 53.7 ... ..$ slope : num [1:50] 0.017 -0.012 -0.015 0.087 -0.006 0.05 0.076 -0.001 0.056 0.042 ... ..$ MSPE : num [1:50] 319 153 203 194 277 ... $ theta :'data.frame': 50 obs. of 2 variables: ..$ G: num [1:50] 27.3 29.2 30.8 24.6 29.5 ... ..$ E: num [1:50] 138 150 145 150 140 ... $ yobs : num [1:1500] 26.6 37.9 40.6 50.1 47.8 ... $ ypred : num [1:1500] 53.2 55.8 44.9 44.9 58.7 ... library(knitr) library(kableExtra) kable(cvG$accuracy[1:5,], caption = "Cross Validation Predictive Ability") %>% kable_styling(full_width = F, position = "left")
交叉验证预测能力
Corr R2 Nagel R2 AUC intercept slope MSPE
0.087 0.008 NA NA 50.840 0.017 319.024
-0.049 0.002 NA NA 52.728 -0.012 152.879
-0.058 0.003 NA NA 54.540 -0.015 202.904
0.388 0.150 NA NA 49.077 0.087 194.324
-0.026 0.001 NA NA 53.673 -0.006 276.643
kable(cvG$theta[1:5,], caption = "Cross Validation Parameter Estimates") %>% kable_styling(full_width = F, position = "left") 交叉验证参数估计
G -------- E
27.278 138.422
29.239 150.142
30.825 144.888
24.614 150.377
29.532 140.285
GREML交叉验证分析的输出
输出包括量化模型预测能力的统计数据,通过回归验证数据集观察到的表型与预测的表型进行评估:
y=intercept+y^slope+e
cvG$accuracy中值的解释:
| Value | Description |
|---|---|
| Corr | 预测表型值与观察表型值的相关性。平均列出的50个相关关系产生了预测能力 |
| R2 | R2, GBLUP模型所能解释的总方差的比例 |
| – | – |
| Nagel R2 | Nagelkerke’s R |
| AUC | ROC曲线下面积 |
| – | – |
| intercept | y回归到y^的y轴截距 |
| slope | y回归到y^的斜率 |
| – | – |
| MSPE | 均数平方预测误差= 1/nv∑nvi=1 (yi−yi^)2, nv =验证集观测数 |
cvG$theta值的解释:

准备结果的数据框架。
讯享网accuMeans <- colMeans(cvG$accuracy) thetaMeans <- colMeans(cvG$theta) accuSEM <- apply(cvG$accuracy, 2, function(x){sd(x)/sqrt(50)}) thetaSEM <- apply(cvG$theta, 2, function(x){sd(x)/sqrt(50)}) results1 <- round( rbind(accuMeans, accuSEM), digits=3) results2 <- round( rbind(thetaMeans, thetaSEM), digits=3) results <- cbind(c("mean","sem"),results1,results2) rownames(results) <- NULL kable(results, caption = "Results Summary")%>% kable_styling(position = "left")
结果摘要

遗传率(h 2)
h2fm <- cvG$theta$G/(cvG$theta$G+cvG$theta$E) boxplot(h2fm, main = "Genomic Heritability") 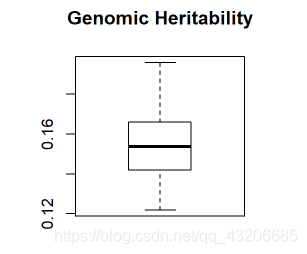
遗传值直方图
讯享网hist(fitG$g, xlab = "genetic value", cex.lab=0.8, cex.axis=0.8, main = "REML", cex.main=0.8)

表型的柱状图
hist(y, xlab = "hours", cex.lab=0.8, cex.axis=0.8, main = "Starvation Resistance", cex.main=0.8) 
二、使用GFBLUP和交叉验证进行预测
这里,我们将展示如何使用qgg中的greml函数执行基因组特征**线性无偏预测(GFBLUP)分析和交叉验证。GFBLUP模型包括一个额外的基因组效应,量化了一组标记的集体行为,即一个基因组特征。在这个例子中,与染色体相关的标记被认为是一个特征(标记集)。GFBLUP的执行涉及使用限制最大似然估计(REML)估计每个特征集的方差分量。GFBLUP模型预测表型的能力通过交叉验证程序进行评估。这将通过Drosophil提供的“抗饥饿”表型和基因型数据来说明
要执行GFBLUP分析,以下输入数据是必不可少的。
y : vector of phenotype
X: design matrix for covariables
Genomic feature marker sets
W: centered and scaled genotype matrix
G: genomic relationship matrix for each feature
该脚本包括以下步骤:
1)加载和准备数据
2)用于估计方差成分的限制最大似然(REML)分析
3)使用GFBLUP模型进行REML分析和交叉验证
讯享网#library(devtools) #install_github("psoerensen/qgg") library(qgg)
加载并准备用于GFBLUP分析的数据
加载抗饥饿数据的已清理和编辑的数据框。
load(file = "./phenotypes/starv_inv_wo.Rdata") dim(starv) load(file = "./phenotypes/starv_inv_wo.Rdata") dim(starv) 创建一个抗饥饿表型的向量y。
讯享网data <- starv y <- data$y length(y) [1] 406
为共变量准备设计矩阵X。fm是设计矩阵中包含相关协变量的公式。
fm <- y ~ wo + In2Lt + In2RNS + In3RP + In3RK + In3RMo X <- model.matrix(fm, data=data) X[1:5,] (Intercept) woy In2LtINV/ST In2LtST In2RNSINV/ST In2RNSST In3RPINV/ST 1 1 1 1 0 0 1 0 2 1 0 1 0 0 1 0 3 1 0 0 1 0 1 0 4 1 0 1 0 0 1 0 5 1 0 0 1 0 1 0 In3RPST In3RKINV/ST In3RKST In3RMoINV/ST In3RMoST 1 1 0 0 0 1 2 1 0 1 0 1 3 1 0 0 0 1 4 1 0 1 0 1 5 1 0 1 0 1 加载集中和缩放的基因型矩阵W。
讯享网load(file = "./genotypes/dgrp2_W2.Rdata") dim(W) [1] 205 W[1:5,1:5] 2L_5317 2L_5372 2L_5390 2L_5403 2L_5465 21 -0. -0. 1. -0. -0. 26 -0. -0. -0. -0. -0. 28 -0. -0. 1. -0. -0. 31 4.0006648 -0. -0. -0. -0. 32 -0. -0. -0. -0. -0.
load(file = "./annotation/chrSets2.Rdata") str(chrSets) List of 6 $ 2L: chr [1:] "2L_5317" "2L_5372" "2L_5390" "2L_5403" ... $ 2R: chr [1:] "2R_10037" "2R_10468" "2R_10959" "2R_12079" ... $ 3L: chr [1:] "3L_39998" "3L_40145" "3L_40202" "3L_40635" ... $ 3R: chr [1:] "3R_1339" "3R_1651" "3R_2158" "3R_2318" ... $ 4 : chr [1:2686] "4_61790" "4_62622" "4_62905" "4_62908" ... $ X : chr [1:] "X_19380" "X_19797" "X_20390" "X_20491" ... 只保留W矩阵中的snp。
讯享网setsGF <- lapply(chrSets,function(x){ x[x%in%colnames(W)] })
创建对象nsets:集合的数量(染色体的数量)和nsnps:每条染色体的SNPs数量。
nsets <- length(setsGF) nsnps <- sapply(setsGF,length) 加性基因组关系矩阵G (VanRaden PM)。2008. 利用所有遗传标记G=WW ’ /m,其中W为中心和缩放的基因型矩阵,m为标记总数。基因组特征关系矩阵(GF) Gk=WkW 'k /mk,为第k个遗传标记集的加性基因组关系矩阵。
从基因型矩阵W计算累加的基因组关系矩阵G。
讯享网L <- data$L GF <- lapply(setsGF, function(x) {grm(W = W[L, x])})
测定了每个系雄性和雌性的抗饥饿能力。因此GF每条染色体有406行和406列的维度。
str(GF) List of 6 $ 2L: num [1:406, 1:406] 0.50583 0.09563 -0.00765 0.11707 -0.03977 ... ..- attr(*, "dimnames")=List of 2 .. ..$ : chr [1:406] "100" "101" "105" "109" ... .. ..$ : chr [1:406] "100" "101" "105" "109" ... $ 2R: num [1:406, 1:406] 0. -0.000345 -0.000574 -0.005291 -0.005337 ... ..- attr(*, "dimnames")=List of 2 .. ..$ : chr [1:406] "100" "101" "105" "109" ... .. ..$ : chr [1:406] "100" "101" "105" "109" ... $ 3L: num [1:406, 1:406] 1.0584 -0.0089 0.0159 0.0062 0.0072 ... ..- attr(*, "dimnames")=List of 2 .. ..$ : chr [1:406] "100" "101" "105" "109" ... .. ..$ : chr [1:406] "100" "101" "105" "109" ... $ 3R: num [1:406, 1:406] 1.4228 -0.0146 0.3536 -0.0101 -0.0248 ... ..- attr(*, "dimnames")=List of 2 .. ..$ : chr [1:406] "100" "101" "105" "109" ... .. ..$ : chr [1:406] "100" "101" "105" "109" ... $ 4 : num [1:406, 1:406] 0.599 -0.254 -0.319 0.239 0.108 ... ..- attr(*, "dimnames")=List of 2 .. ..$ : chr [1:406] "100" "101" "105" "109" ... .. ..$ : chr [1:406] "100" "101" "105" "109" ... $ X : num [1:406, 1:406] 1.01855 0.00487 0.02768 0.0022 -0.01247 ... ..- attr(*, "dimnames")=List of 2 .. ..$ : chr [1:406] "100" "101" "105" "109" ... .. ..$ : chr [1:406] "100" "101" "105" "109" ... 限制最大似然(REML)分析
greml函数返回一个列表结构,其中包括对固定效应(b)、随机效应(yk)、剩余效应(e^)的估计,以及方差成分(xyc2gk)。列表中的其他值在greml帮助页面中描述。将verbose = FALSE更改为verbose = TRUE,可以查看迭代。略…
REML使用GFBLUP模型进行分析和交叉验证
从训练数据(t,365行)中估计表型的基因组参数,从验证数据(v, 41行)中预测表型。根据训练和验证群体之间的基因组关系来预测遗传价值:

k^th的基因组关系矩阵

根据训练数据中的个体之间的关系Gttk,验证数据中的个体之间的关系Gvvk,以及训练数据和验证数据中的个体之间的关系Gvtk进行划分。因此,使用估计的方差组成部分来预测总基因组易感性(utils2g1,…在训练数据中,如:eurch2nf和eurch2e)。最右边的术语(yt−Xtbt)构成了训练数据中个体为固定效应而校正的表型。反项V - 1t本质上是校正后的表型的方差-协方差结构。这两个术语相乘就是训练数据中个体的标准化和校正的表型,这些表型被投射到训练数据和验证数据之间的总遗传协方差结构上。

通过估计训练集的方差分量,预测验证集的表型为:

5验证集是通过从1 - 406 (y的长度)中随机抽样41个值创建的,并重复这个抽样5次。验证集保存在验证矩阵中。这个矩阵指定GREML分析中要使用的数据行。
由于染色体2L和3L的方差分量为0,因此只有染色体2R和3R作为y预测的特征。
讯享网n <- length(y) validate <- replicate(5, sample(1:n, 41)) cvG <- greml(y = y, X = X, GRM = GF[c(2, 4)], validate = validate) str(cvG) library(knitr) library(kableExtra) kable(cvG$accuracy[1:5,], caption = "Cross Validation Predictive Ability") %>% kable_styling(full_width = F, position = "left")
交叉验证预测能力

kable(cvG$theta[1:5,], caption = "Cross Validation Parameter Estimates") %>% kable_styling(full_width = F, position = "left") 交叉验证参数估计

GREML交叉验证分析的输出
准备结果的数据框架。
讯享网accuMeans <- colMeans(cvG$accuracy) thetaMeans <- colMeans(cvG$theta) accuSEM <- apply(cvG$accuracy, 2, function(x){sd(x)/sqrt(50)}) thetaSEM <- apply(cvG$theta, 2, function(x){sd(x)/sqrt(50)}) results1 <- round( rbind(accuMeans, accuSEM), digits=3) results2 <- round( rbind(thetaMeans, thetaSEM), digits=3) results <- cbind(c("mean","sem"),results1,results2) rownames(results) <- NULL kable(results, caption = "Results Summary")%>% kable_styling(position = "left")
结果摘要

基因组遗传力
theta <- cvG$theta h2f2 <- theta["2R"]/sum(theta) h2f4 <- theta["3R"]/sum(theta) layout(matrix(1:2,ncol=2)) boxplot(h2f2, main = "Genomic Heritability 2R", cex.main=0.8) boxplot(h2f4, main = "Genomic Heritability 3R", cex.main=0.8) 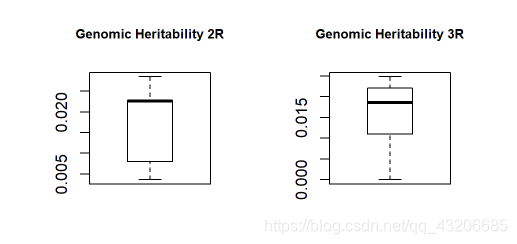
遗传值直方图
讯享网layout(matrix(1:2,ncol=2)) hist(fitGF$g[,2], xlab = "genetic value", cex.lab=0.8, cex.axis=0.8, main = "REML, chromosome 2R", cex.main=0.8) hist(fitGF$g[,4], xlab = "genetic value", cex.lab=0.8, cex.axis=0.8, main = "REML, chromosome 3R", cex.main=0.8)

表型的柱状图
hist(y, xlab = "hours", cex.lab=0.8, cex.axis=0.8, main = "Starvation Resistance", cex.main=0.8) 
三、基因集富集分析
在本文中,一系列不同的基因集富集分析方法将通过果蝇遗传参考面板(DGRP)提供的“抗饥饿”表型和基因型数据来说明。
qgg中的gsea功能可以进行四种不同的基因集富集分析。在这里,我们展示如何执行我们自己开发的协方差关联测试(CVAT),基于分数的基于求和和基于计数的(hyperG)基因集富集分析。有关试验统计数据的详细信息和比较,请查阅doi:10.1534/genetics.116.。
一般的程序是获得单一的标记统计(例如总结统计),从它可以计算和评估一组遗传标记的测试统计,以衡量标记集和表现型之间的联合程度。标记集由基因、生物途径、基因相互作用、基因表达谱等基因组特征来定义。
要进行不同类型的基因集富集分析,以下输入数据是必不可少的。
y : vector of phenotype
X: design matrix for covariables
Genomic feature marker sets
W: centered and scaled genotype matrix
G: genomic relationship matrix for each feature
gblup衍生的标记集检验方法基于三个步骤:首先拟合一个线性混合模型,然后计算单个标记的效果,最后计算标记集(基因组特征)的检验统计量。
该脚本包括以下步骤:
1)加载和准备数据,
2)GBLUP线性混合模型,限制性最大似然分析,
3)CVAT,
4)基于分数的,
5)基于累加的,
6)基于计数的基因集富集分析。
讯享网#library(devtools) #install_github("psoerensen/qgg") library(qgg)
加载和准备数据
加载抗饥饿数据的已清理和编辑的数据框。
load(file = "./phenotypes/starv_inv_wo.Rdata") dim(starv) [1] 406 20 head(starv) L sex y In2Lt In2RNS In2RY1 In2RY2 In2RY3 In2RY4 In2RY5 In2RY6 1 100 M 49.28000 INV/ST ST ST ST ST ST ST ST 2 101 M 47.20000 INV/ST ST ST ST ST ST ST ST 3 105 M 51.04000 ST ST ST ST ST ST ST ST 4 109 M 44.96000 INV/ST ST ST ST ST ST ST ST 5 129 M 33.08475 ST ST ST ST ST ST ST ST 6 136 M 63.04000 ST ST ST ST ST ST ST ST In2RY7 In3LP In3LM In3LY In3RP In3RK In3RMo In3RC wo 1 ST ST ST ST ST INV ST ST y 2 ST ST ST ST ST ST ST ST n 3 ST ST ST ST ST INV ST ST n 4 ST ST ST ST ST ST ST ST n 5 ST ST ST ST ST ST ST ST n 6 ST INV/ST ST ST ST INV/ST ST ST y 创建一个抗饥饿表型的向量y。
讯享网data <- starv y <- data$y length(y) [1] 406
为共变量准备设计矩阵X。fm是设计矩阵中包含相关协变量的公式。
fm <- y ~ wo + In2Lt + In2RNS + In3RP + In3RK + In3RMo X <- model.matrix(fm, data=data) X[1:5,] (Intercept) woy In2LtINV/ST In2LtST In2RNSINV/ST In2RNSST In3RPINV/ST 1 1 1 1 0 0 1 0 2 1 0 1 0 0 1 0 3 1 0 0 1 0 1 0 4 1 0 1 0 0 1 0 5 1 0 0 1 0 1 0 In3RPST In3RKINV/ST In3RKST In3RMoINV/ST In3RMoST 1 1 0 0 0 1 2 1 0 1 0 1 3 1 0 0 0 1 4 1 0 1 0 1 5 1 0 1 0 1 加载集中和缩放的基因型矩阵W。
讯享网load(file = "./genotypes/dgrp2_W2.Rdata") dim(W) [1] 205 W[1:5,1:5] 2L_5317 2L_5372 2L_5390 2L_5403 2L_5465 21 -0. -0. 1. -0. -0. 26 -0. -0. -0. -0. -0. 28 -0. -0. 1. -0. -0. 31 4.0006648 -0. -0. -0. -0. 32 -0. -0. -0. -0. -0.
基于基因本体(GO)加载SNP集、goset。
load(file="./annotation/goSets2.Rdata") str(goSets[1:5]) List of 5 $ GO:0000022: chr [1:315] "2R_" "2R_" "2R_" "2R_" ... $ GO:0000027: chr [1:644] "X_" "X_" "X_" "X_" ... $ GO:0000028: chr [1:283] "X_" "X_" "X_" "X_" ... $ GO:0000045: chr [1:1602] "X_" "X_" "X_" "X_" ... $ GO:0000060: chr [1:1816] "X_" "X_" "X_" "X_" ... 只保留W矩阵中的snp。
讯享网goSets <- lapply(goSets,function(x){ x[x%in%colnames(W)] })
创建对象nset:集合的数量和nsnp(每个GO项的snp数量)。
nsets <- length(goSets) nsnps <- sapply(goSets,length 加性基因组关系矩阵G (VanRaden PM)。2008. 利用所有遗传标记G=WW ’ /m,其中W为中心和缩放的基因型矩阵,m为标记总数。
从基因型矩阵W计算累加的基因组关系矩阵G。
讯享网L <- data$L G <- grm(W = W) G <- G[L,L]
测定了每个系雄性和雌性的抗饥饿能力。因此G的维数是406行和406列。
str(G) num [1:406, 1:406] 1.0327 0.006 0.0889 0.0128 -0.0133 ... - attr(*, "dimnames")=List of 2 ..$ : chr [1:406] "100" "101" "105" "109" ... ..$ : chr [1:406] "100" "101" "105" "109" ... GBLUP线性混合模型
GBLUP基于线性混合模型,仅包含一种随机基因组效应:
y=Xb+Zg+e
其中y为表型观察向量,X和Z为固定效应和随机效应的设计矩阵,b为固定效应向量,g为所有遗传标记捕获的基因组值向量,e为残差向量。随机基因组值和残差被假设为独立的正态分布值,描述如下:g ~ N(0, g ggg2g)和e ~ N(0,I = bx2e)。因此,我们假设观察到的表型为y ~ N(Xb,V),其中V=ZGZ ’ g2g +I g2e。
限制最大似然(REML)分析
使用REML分析(greml函数)估计方差分量,对于用于GBLUP分析的线性混合模型中的随机效应,分别为两个型号的2g和2e。
根据在整个研究群体中观察到的表型来预测遗传价值:

表型预测为:

greml函数在收敛之前要经过多次迭代(即,连续轮之间的参数变化小于指定的阈值,请参阅greml帮助页面中的“tol”参数)。在本例中,每次迭代都返回方差组件(第3列)和(第4列)的值.
greml函数返回一个列表结构,其中包括对固定效果(b)、随机效果(g)和剩余效果(e)的估计。列表中的其他值在greml帮助页面中描述。
讯享网fit <- greml(y=y, X=X, GRM=list(G=G), verbose = FALSE)
单一标记的效应
单一标记效应的计算建立在gsea函数中。
单标记效应s^由GBLUP模型预测的总基因组值

计算得出。单标记效果计算如下:

计算出单标记效应的(co)方差为:

个体标记与表型的联系是基于单一标记测试统计,如t统计和该统计的阈值。

其中Var(s^j)是由单标记效应的(co)方差矩阵对角的第j个元素得到的s^的第j个元素的方差估计。
零假设下,s^j=0,假设ts^j服从dfe剩余自由度的分布.(for further information, see Sørensen IF. et al., 2017. Scientific Reports. doi:10.1038/s41598-017-02281-3).
基因集合富集分析
在本例中,与GO项相关的遗传标记被认为是一个标记集(基因组特征)。因此,在这种情况下,基因组特征是由GO项目定义的。这里我们描述了四种不同的集合测试方法,它们都来自于GBLUP模型。
这包括
1)协方差协会组测试(CVAT),
2)基于评分的方法,
3)测试统计基于遗传标记的总和属于同一基因的功能,和
4)基于计算检验统计量的数量遗传标记的功能与特征相关的表型(hyperG)。
在这个例子中,我们考虑了一个竞争性零假设,也就是说,相关的标记是从被测试遗传标记的总数中随机挑选出来的。
协方差关联检验统计量(Tcvat)
在这里,我们考虑所有标记的总基因组效应和基因组特征中遗传标记集的基因组效应之间的协方差


在这个表达式,g^r=∑mr i=1 wis^i ,是剩余标记的基因组效应,例如,不包括在特性中的标记。mf和mr分别为feature中标记的数量和剩余的标记集。TCVAT也与解释的遗传标记的平方和有关,该检验统计量在原假设下的分布很难用精确或近似的分布来描述,需要经验分布。通过对W中的mf列随机抽样,可以得到竞争零假设的经验分布。
gsea函数中的fit参数是使用greml函数对线性混合模型进行拟合得到的fit对象。
gsea函数返回一个数据框架(mma,多标记关联对象),其中包括协方差测试统计量(setT)、集合中的标记数(g)和p值§。
mma <- gsea(fit = fit, sets = goSets, W = W, method = "cvat") 基于评分的方法统计(Tscore)
它是由似然的一阶导数推导出来的,就像Rao的分数测试一样(Rao et al. 1948)。比饶的分数测试关键的区别是,只有一阶导数的二次项的基础测试统计(Goeman et al. 2004, Wu et al. 2011, Wang et al. 2013 )从一个论点,这是唯一的一部分,涉及到数据(Huang and Lin, 2013)。因此,这里给出的基于分数的方法相当于序列核关联测试(SKAT, Wu et al. 2011)。因此得分统计量可以写成:

其中固定效应b和表型协方差矩阵V在零模型下估计。空模型的目的是为了调整环境的非遗传因素,遗传因素不是基因组特征的一部分,包括群体结构。对于GBLUP零模型,基因组效应可以定义为g ~ N(0, g ggg2g),也可以定义为g ~ N(0,Gr Gr Gr Gr 2r) (r = remaining)。在第一种情况下,基因组关系矩阵是使用所有遗传标记计算,因此null模型只需要拟合一次。在后一种情况下,只使用不包括在基因组特征中的遗传标记进行计算,这需要为每个基因组特征拟合不同的模型。
得分法的测试统计量集合可以改写为:


通过对W中的mf列随机抽样,得到了在竞争零假设下得分检验统计量的经验分布。
gsea函数返回一个数据框架(mma),其中包括协方差测试统计量(stat),集合中标记数(g)和p值§。
讯享网mma <- gsea(fit = fit, sets = goSets, W = W, method = "score")
基于总和(Tsum)设置检验统计量
这个总和集测试是基于位于特征集中的所有标记汇总统计量的测试统计量之和,例如:
其中ti表示第i个单变量检验统计量,如标记效应(s^)或t统计量。可以从线性模型分析(从PLINK或使用qgg lma近似)中获得单一标记汇总统计信息,或者greml函数中的单个或多个组件REML分析(GBLUP或GFBLUP)。如果基因组特征含有许多具有小到中度影响的遗传标记,那么总和检验是强有力的。该检验统计量在竞争零假设下的分布是未知的,需要一个经验分布。s^和t统计量都用来计算Tsum。
基于sum的set测试可以由gsea函数执行,并将方法参数指定为“sum”。
ma <- lma(y=y,X=X,W=W[L,]) mma <- gsea(stat = ma[,"stat"]2, sets = goSets, method = "sum") head(mma) m stat p GO:0000022 315 424.3648 0.253 GO:0000027 644 921.7163 0.145 GO:0000028 283 307.9064 0.466 GO:0000045 1602 1659.4118 0.583 GO:0000060 1816 1587.1936 0.881 GO:0000070 3416 3458.3678 0.703 基于hyperG计算测试统计量(Tcount)
该检验统计量是通过对特征中与性状表型相关的遗传标记的数量进行计数,计算方法为:

其中mf为特征中标记数,ti为第i个单标记检验统计量(如t统计量),t0为单标记检验统计量的任意选择阈值,I是和指示器函数,如果满足参数(abs(ti)>t0),它将取值1。在零假设(即个体相关的标记随机分布)下,假设Tcount ~ Hyper(m,ma,mf)是一个参数为m的超几何分布(测试的遗传标记总数)的实现,ma(所有标记中相关遗传标记的总数)和mf(特征中遗传标记的总数)。gsea函数可以执行基于sum的set测试,并将方法参数指定为“hyperg”。
讯享网ma <- lma(y=y,X=X,W=W[L,]) head(ma) s se stat p 2L_5317 0. 0. 0. 0. 2L_5372 0. 0. 0. 0. 2L_5390 0. 0. 0. 0. 2L_5403 0. 0. 0. 0. 2L_5465 0. 0. 0. 0. 2L_5598 -0. 0. -0. 0. mma <- gsea(stat = ma[,"p"], sets = goSets[1:10], method = "hyperg", threshold = 0.05) head(mma) GO:0000022 GO:0000027 GO:0000028 GO:0000045 GO:0000060 5.036082e-02 3.e-06 9.e-01 9.e-01 9.e-01 GO:0000070 9.e-01
结果表明,每组snp与表型之间的显著性相关的p值。
版权声明:本文内容由互联网用户自发贡献,该文观点仅代表作者本人。本站仅提供信息存储空间服务,不拥有所有权,不承担相关法律责任。如发现本站有涉嫌侵权/违法违规的内容,请联系我们,一经查实,本站将立刻删除。
如需转载请保留出处:https://51itzy.com/kjqy/125752.html