
由于受目前测序水平的限制,基因组测序时需要先将基因组打断成DNA片段,然后再建库测序。reads(读长)指的是测序仪单次测序所得到的碱基序列,也就是一连串的ATCGGGTA之类的,它不是基因组中的组成。不同的测序仪器,reads长度不一样。对整个基因组进行测序,就会产生成百上千万的reads。
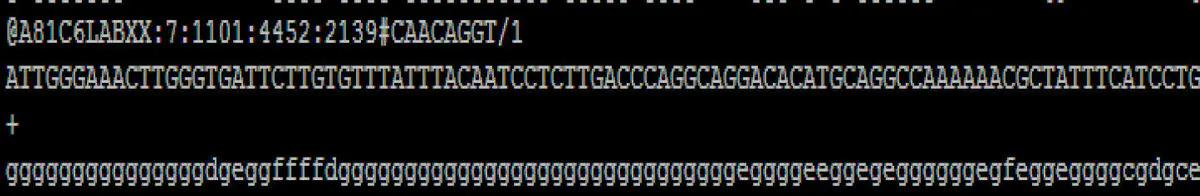
测序得到的原始图像数据经 base calling 转化为序列数据,我们称之为 raw data 或 raw reads ,结果以 fastq 文件格式存储, fastq 文件为用户得到的最原始文件,里面存储 reads 的序列以及 reads 的测序质量。在 fastq 格式文件中每个 read 由四行描述:
@read ID TGGCGGAGGGATTTGAACCC + bbbbbbbbabbbbbbbbbbb
讯享网
- Single-end(SE)测序:1个fastq文件
- Pair-end(PE)测序:2个fastq文件分别存放read1和read2的数据
讯享网Q = -10 log10P
Solexa测序错误率与测序质量值简明对应关系:


高通量测序时,在芯片上的每个反应,会读出一条序列,是比较短的,叫read,它们是原始数据;
有很多reads通过片段重叠,能够组装成一个更大的片段,称为contig;
多个contigs通过片段重叠,组成一个更长的scaffold;
版权声明:本文内容由互联网用户自发贡献,该文观点仅代表作者本人。本站仅提供信息存储空间服务,不拥有所有权,不承担相关法律责任。如发现本站有涉嫌侵权/违法违规的内容,请联系我们,一经查实,本站将立刻删除。
如需转载请保留出处:https://51itzy.com/kjqy/53901.html