
想用GEO里面的数据找一种皮肤病的差异基因,有三组数据:正常人皮肤,病人皮损皮肤和病人非皮损皮肤。在进行病人皮损与非皮损两组之间比较的时候用配对t检验,在进行这两组分别与正常人皮肤比较时候要用成组t检验,有什么分析软件可以实现呢?如果用limma R包可以吗,写程序时候有什么区别?
今天做一下《journal of extracellular vesicles》IF:25上的一个图,其实图不是重点,重要的是这个思路。

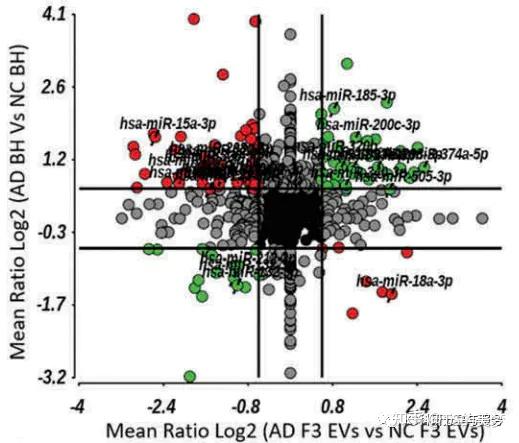
做这个图需要的数据是两组差异比较的差异倍数和FDR,这里我们首先用ggplot画一个散点图。
library(ggplot2) library(ggrepel) ggplot(A, aes(x=MeanRatio_X, y=MeanRatio_Y)) + geom_hline(yintercept= c(-0.6, 0.6), color = “black”, size=1) +#添加横线 geom_vline(xintercept=c(-0.6, 0.6), color = “black”, size=1)+ geom_point() 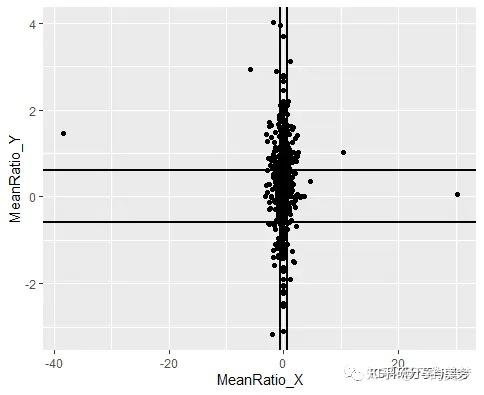
可以看到x轴y轴范围和文章不一样,这里不清楚作者为什么这么设置,暂且按照它的数据吧。接下来需要修饰坐标轴,标记不同象限的点。
GPT plus 代充 只需 145ggplot(A, aes(x=MeanRatio_X, y=MeanRatio_Y)) + geom_hline(yintercept= c(-0.6, 0.6), color = “black”, size=1) + geom_vline(xintercept=c(-0.6, 0.6), color = “black”, size=1)+ geom_point(size=3)+ geom_point(data = A[A\(MeanRatio_X>0.6 | A\)MeanRatio_X < -0.6, ], size=3, shape=16, color=“grey”)+ geom_point(data = A[A\(MeanRatio_Y>0.6 | A\)MeanRatio_Y < -0.6, ], size=3, shape=16, color=“grey”)+ geom_point(data = A[A\(MeanRatio_X>0.6 & A\)MeanRatio_Y>0.6, ], size=3, shape=16, color=“#66CC33”)+ geom_point(data = A[A\(MeanRatio_X< -0.6 & A\)MeanRatio_Y< -0.6, ], size=3, shape=16, color=“#66CC33”)+ geom_point(data = A[A\(MeanRatio_X< -0.6 & A\)MeanRatio_Y > 0.6, ], size=3, shape=16, color=“#CC3300”)+ geom_point(data = A[A\(MeanRatio_X > 0.6 & A\)MeanRatio_Y < -0.6, ], size=3, shape=16, color=“#CC3300”)+ geom_point(size = 3,shape=21)+ xlim(-4,4)+ ylim(-3.2, 4.1)
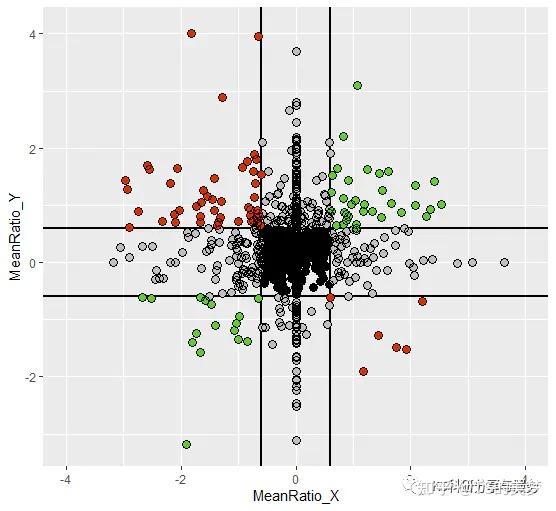
接下来就是修改ggplot主题了。
ggplot(A, aes(x=MeanRatio_X, y=MeanRatio_Y)) + geom_hline(yintercept= c(-0.6, 0.6), color = “black”, size=1) + geom_vline(xintercept=c(-0.6, 0.6), color = “black”, size=1)+ geom_point(size=3)+ geom_point(data = A[A\(MeanRatio_X>0.6 | A\)MeanRatio_X < -0.6, ], size=3, shape=16, color=“grey”)+ geom_point(data = A[A\(MeanRatio_Y>0.6 | A\)MeanRatio_Y < -0.6, ], size=3, shape=16, color=“grey”)+ geom_point(data = A[A\(MeanRatio_X>0.6 & A\)MeanRatio_Y>0.6, ], size=3, shape=16, color=“#66CC33”)+ geom_point(data = A[A\(MeanRatio_X< -0.6 & A\)MeanRatio_Y< -0.6, ], size=3, shape=16, color=“#66CC33”)+ geom_point(data = A[A\(MeanRatio_X< -0.6 & A\)MeanRatio_Y > 0.6, ], size=3, shape=16, color=“#CC3300”)+ geom_point(data = A[A\(MeanRatio_X > 0.6 & A\)MeanRatio_Y < -0.6, ], size=3, shape=16, color=“#CC3300”)+ geom_point(size = 3,shape=21)+ xlim(-4,4)+ ylim(-3.2, 4.1)+ labs(x = “Mean Ratio Log2(AD F3 EVs vs NC F3 EVs)”,GPT plus 代充 只需 145 y = "Mean Ratio Log2(AD BH vs NC BH)", title = "") +
theme(panel.grid = element_blank(),
axis.line = element_line(colour = 'black', size = 1), panel.background = element_blank(), plot.title = element_text(size = 14, hjust = 0.5), plot.subtitle = element_text(size = 14, hjust = 0.5), axis.text = element_text(size = 14, color = 'black'), axis.title = element_text(size = 14, color = 'black'))+ theme(legend.position = “right”)
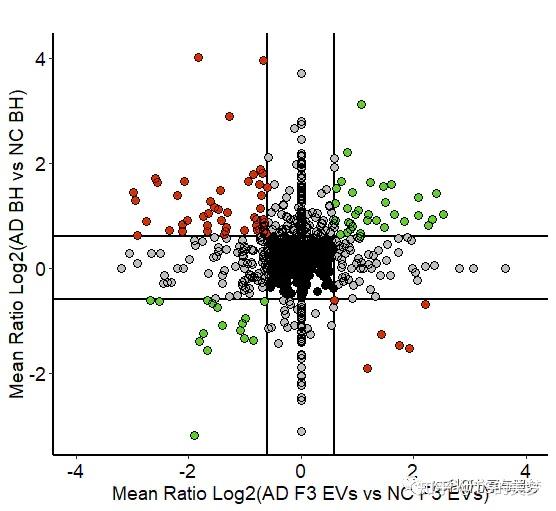
添加标签,原文中作者是选择了一些miRNA呈现,但是呈现效果我认为很差(可能没想着让看),都重叠在一起看不出来,我这里选择显示标签。
GPT plus 代充 只需 145miRNA <- A[which(A\(FDR_Y<0.1 | A\)FDR_X<0.1),] miRNA <- miRNA[which(abs(miRNA\(MeanRatio_Y) >1 |abs(miRNA\)MeanRatio_X)>1),]
ggplot(A, aes(x=MeanRatio_X, y=MeanRatio_Y)) + geom_hline(yintercept= c(-0.6, 0.6), color = “black”, size=1) + geom_vline(xintercept=c(-0.6, 0.6), color = “black”, size=1)+ geom_point(size=3)+ geom_point(data = A[A\(MeanRatio_X>0.6 | A\)MeanRatio_X < -0.6, ], size=3, shape=16, color=“grey”)+ geom_point(data = A[A\(MeanRatio_Y>0.6 | A\)MeanRatio_Y < -0.6, ], size=3, shape=16, color=“grey”)+ geom_point(data = A[A\(MeanRatio_X>0.6 & A\)MeanRatio_Y>0.6, ], size=3, shape=16, color=“#66CC33”)+ geom_point(data = A[A\(MeanRatio_X< -0.6 & A\)MeanRatio_Y< -0.6, ], size=3, shape=16, color=“#66CC33”)+ geom_point(data = A[A\(MeanRatio_X< -0.6 & A\)MeanRatio_Y > 0.6, ], size=3, shape=16, color=“#CC3300”)+ geom_point(data = A[A\(MeanRatio_X > 0.6 & A\)MeanRatio_Y < -0.6, ], size=3, shape=16, color=“#CC3300”)+ geom_point(size = 3,shape=21)+ xlim(-4,4)+ ylim(-3.2, 4.1)+ labs(x = “Mean Ratio Log2(AD F3 EVs vs NC F3 EVs)”,
y = "Mean Ratio Log2(AD BH vs NC BH)", title = "") + theme(panel.grid = element_blank(),
GPT plus 代充 只需 145 axis.line = element_line(colour = 'black', size = 1), panel.background = element_blank(), plot.title = element_text(size = 14, hjust = 0.5), plot.subtitle = element_text(size = 14, hjust = 0.5), axis.text = element_text(size = 14, color = 'black'), axis.title = element_text(size = 14, color = 'black'))+
geom_text_repel(data=miRNA, aes(label=rownames(miRNA)), color=“black”, size=4, fontface=“italic”,
point.padding = 0.3, segment.color = 'black', segment.size = 0.3, force = 1, max.iter = 3e3)
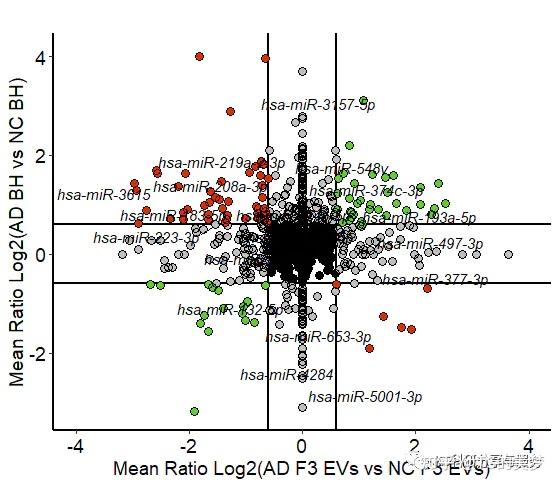
这个图还是很有意义的,显示的信息也挺多。可以结合自己的数据,看能不能有应用的地方。有需要示例数据及注释代码的小伙伴可至我的个人公众号《KS科研分享与服务》!
目 录
GEO数据库-简介
如何找到自己需要数据?
1. 通过在GEO数据库直接检索(类似pubmed的检索) 重点需要看 数据集中每个样本带有的信息:
2. 通过文献搜索
如何整理GEO数据?
高通量数据提取和整理:
模块:差异分析-测序数据-Counts
模块:分组比较图
模块:其他上传模块
芯片数据提取和整理
下载、提取、整理数据
模块:分组比较图(表达差异)
模块:诊断性ROC曲线
模块:生存曲线
模块:其他上传模块
NCBI-GEO主页:https://www.ncbi.nlm.nih.gov/geo/
NCBI-GEO检索页面:https://www.ncbi.nlm.nih.gov/gds/?term=
GEO数据库是最大的公共数据库之一,包含有多种平台的数据,包括常见的芯片数据、高通量数据(RNAseq、ChIP、单细胞测序数据等等…)。由于是开放给作者上传高通量数据,所以即便是同一个平台(或者同一个GPL平台号)的数据中,包含的数据情况也是各式各样的,也可能会存在有数据缺失的情况。
备注:高通量数据很可能没有提供规整的下游数据,具体要看作者上传了什么样的数据。而上游数据的处理对硬实力(分析能力、服务器)要求高。
基本概念
- GSExxx: GSE开头的对应的是数据集的编号
- GSMxxx:GSM开头的对应的是样本编号(一个GSE会含有很多的GSM)
- GPLxxx:GPL开头的对应的是平台编号(一般一个GSE会只含有一个GPL,但是也不排除一个大的GSE含有多个GPL平台的情况)
- GDSxxx: GDS是GEO整理好的芯片数据,这个编号基本可以不用看。
- GEO2R:针对大部分芯片平台的在线差异分析工具。其他高通量数据是没有这个功能的。
1、通过在GEO数据库直接检索
NCBI-GEO检索页面:https://www.ncbi.nlm.nih.gov/gds/?term=
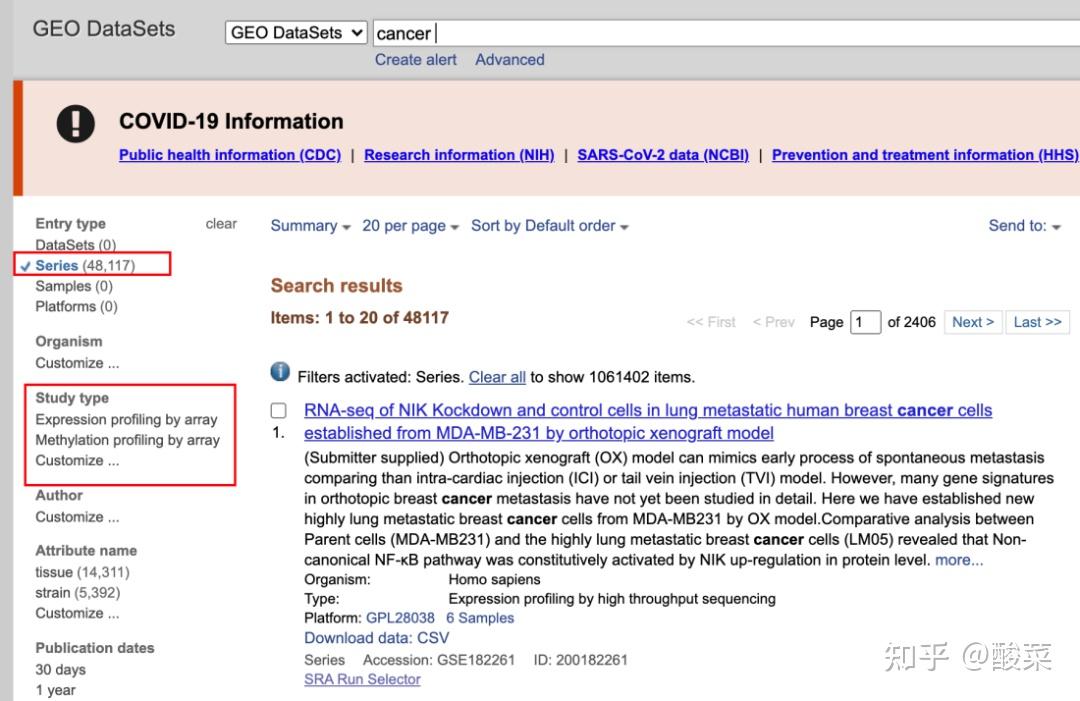
建议右侧限定Series(保证出来的是GSE,不含有GSM(可以不用看GSM))和平台类型(芯片或者RNAseq或者其他)备注:通过GEO数据库搜出来的内容肯定是最全的,任何通过别的数据库或者别的方式搜出来的内容肯定不是最全的。
检索也可以用 仙桃的数据集检索工具(https://www.xiantao.love/gds) 进行:(由于仙桃的数据集检索模块是自建的搜索引擎,所以出来的结果是和GEO上的是不一样的,检索方式也会有不同)
当通过GEO检索数据(并在右侧进行限定)后,出来的结果仍然很多,可以适当再增加一些关键词进行限定,缩小检索的结果。
当检索结果在几十到几百个的时候,可以逐个来检查数据集的设计和分组是否满足自己研究的需要:

首先是简单看看数据集的标题、数据集介绍(可以点进去看具体的介绍和数据集设计和处理情况)
稍微看看 数据集的平台类型(不同)和平台号(有一些平台号会很经常出现,比如GPL570 GPL96等等)
稍重点看看 数据集的样本量(样本量太少重新再纳入意义不大)
重点需要看 数据集中每个样本带有的信息:
如果是高通量数据,需要点进去到数据集介绍页面

样本编号页面都会带有一个关键的信息,通过这个信息基本可以判断这个数据集的样本设计情况。如果样本还含有别的补充信息,需要再点击进入到具体的样本情况页进行查看。
如果是芯片数据,有两种方式进行查看
第一种和高通量数据一样(相对麻烦),一样是点进去到数据集的介绍页查看:
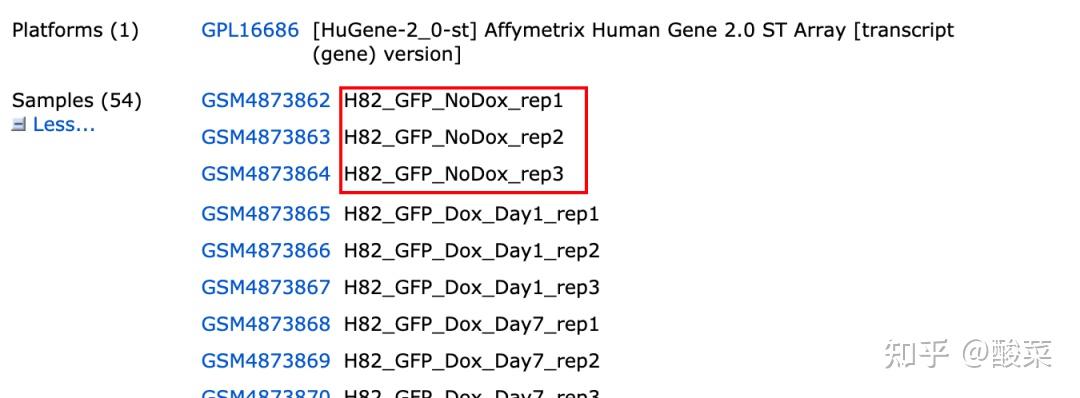
从上面的页面中点击GSM编号跳转到样本介绍页面:
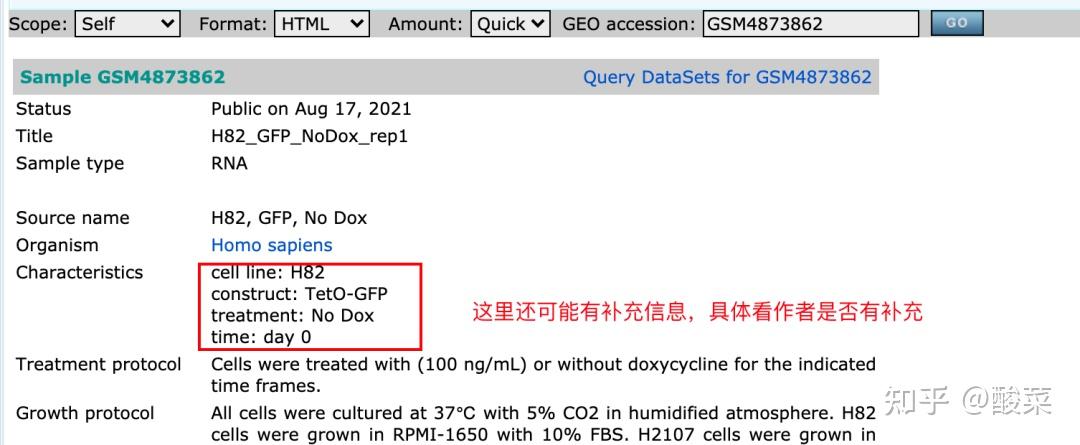
第二种方式:当如果芯片数据有GEO2R的功能:

点击进入到GEO2R,等待数据加载好后,能看到每个样本的补充信息(这部分信息和第一种方式看到的内容基本是一致的,通过GEO2R就不用点开每个样本来查看)
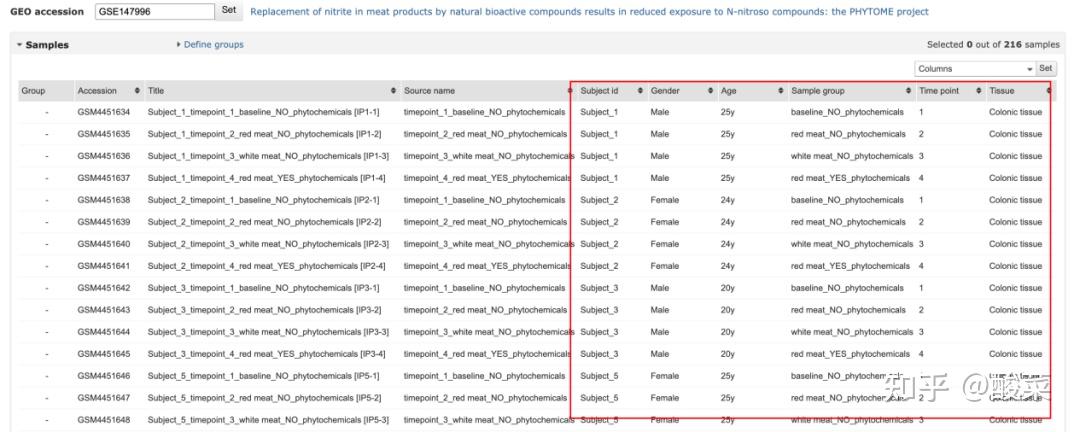
通过GEO2R,能更快速的判断这个数据集中是否 含有自己研究所需要的内容(分组、处理、变量、是否含有预后信息等等)
第三种方式,在数据集介绍页面,下载Series_matrix文件:
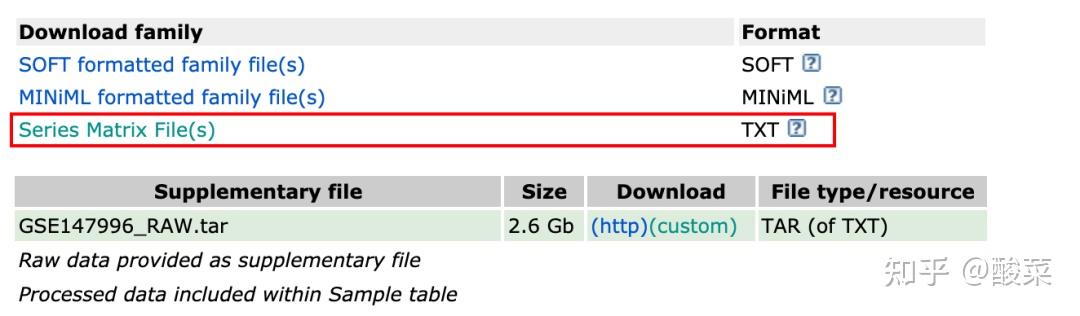
解压后用excel打开txt文件:
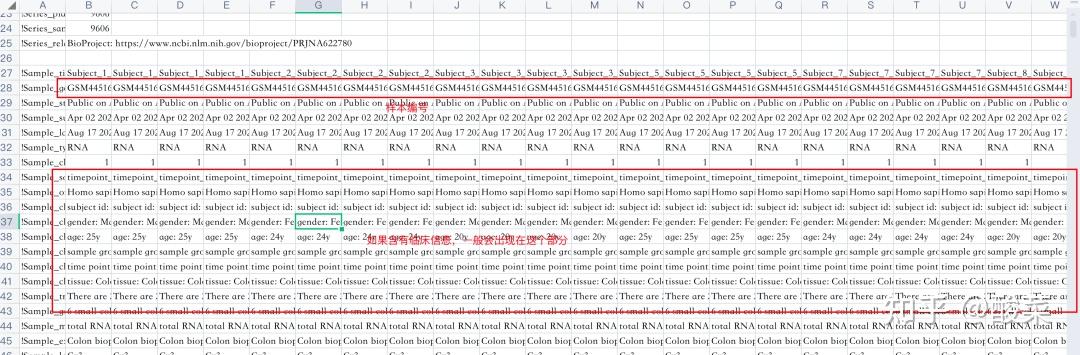
在文件的头部,样本编号的下方,如果作者有提供每个样本的补充信息,则会出现在上图中框住的部分。
这部分数据和GEO2R中看到的基本是一样的。这种也是快速获取数据集中临床信息的一种方式。
2、通过文献搜索
也可以通过搜索相同领域内的文章来看文章里面都用了哪些数据集,搜索文章的时候可以带上GEO 或者类似的关键词在pubmed中进行搜索,搜到的文章中带有的数据集都是相对可能比较好比较常用的数据集,而且还能知道这个数据集能用在什么场景(表达、预后等)
但是,这种方法不一定搜的全,而且工作量也不少。
当如果只是想找一两个数据集来验证 分子的表达或者预后的情况,可以通过这种方式来找。
当通过上面的方法找到了合适的GEO数据后,就可以开始提取和整理数据。
1、高通量数据提取和整理
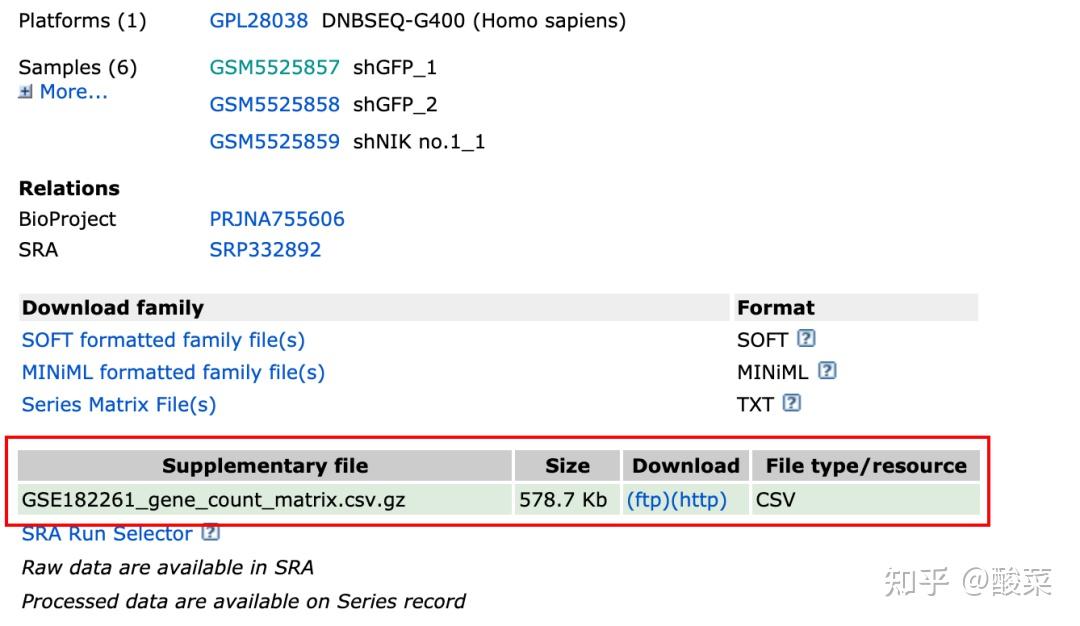
一般高通量的下游数据是在数据集介绍页面的 补充文件中进行下载。(原始上游数据这里不进行介绍,这部分数据不同作者上传的可能差别很大,具体要对应的文件名或者对应的文献介绍确认文件中的数据类型),其他部分的数据可以不用下载(因为这个页面中其他的文件基本没有提供其他有用的信息)
样本信息的内容需要在样本详情页中进行摘录。
模块:差异分析-测序数据-Counts
当对应的数据集含有 原始的counts值(没有进行过转化)的文件时(比如上图),可以下载对应的数据并进行整理(如果不在一个表格,把所有数据放到一个表格中,并在第一行增加分组信息(差异分析比较的两个组)):(数据集的分组数据可以看“如何找到自己需要的数据”这部分的内容中的样本信息部分)
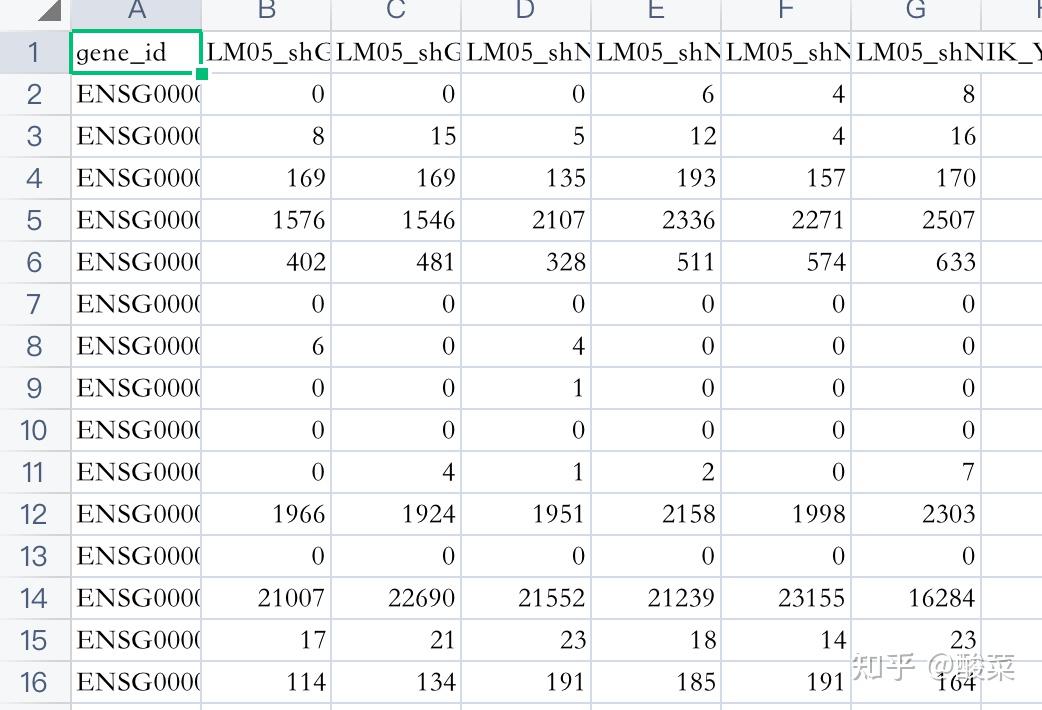
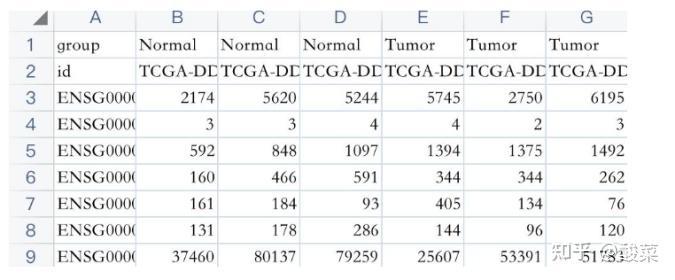
模块:分组比较图
在下载的数据中搜索分子,提取需要的数据成两列(或者多列)(类似下图):(数据集的分组数据可以看“如何找到自己需要的数据”这部分的内容中的样本信息部分)
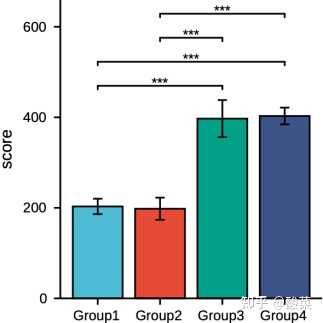
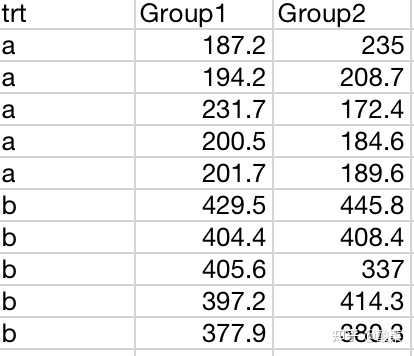
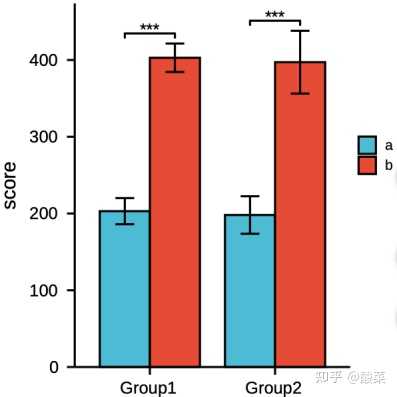
模块:其他上传模块
其他模块也是类型的,只要是上传数据类型的模块,可以通过查看“教程文档”的数据格式说明进行类似的整理数据

1、芯片数据提取和整理
(在这个部分内容时,请保证已经看完了“如何找到自己需要数据”中的芯片数据部分的内容(里面提及了临床和分组信息如何获取的说明))当确定了要用哪些数据集后(或者哪些数据集是有用(含有需要的信息)):(如何确定数据集请看“如何找到自己需要数据”部分)
下载、提取、整理数据
一般需要下载2个文件:
1. 数据集对应的series_matrix(数据集页面)(内部含有表达谱和分组信息(分组信息如何看可以看“如何找到自己需要数据”中的芯片数据部分))
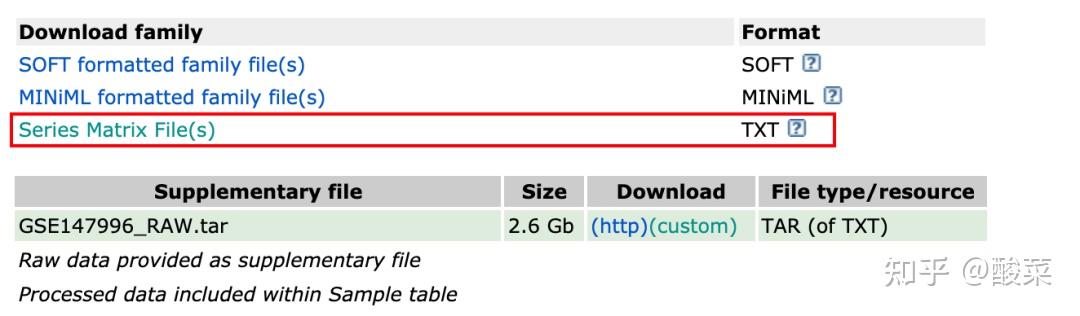
2. 数据集对应的平台文件(内部含有 探针-分子注释信息(部分平台是不含有分子名的注释信息的))

可以在数据集详情页,点击上面的GPL编号跳转
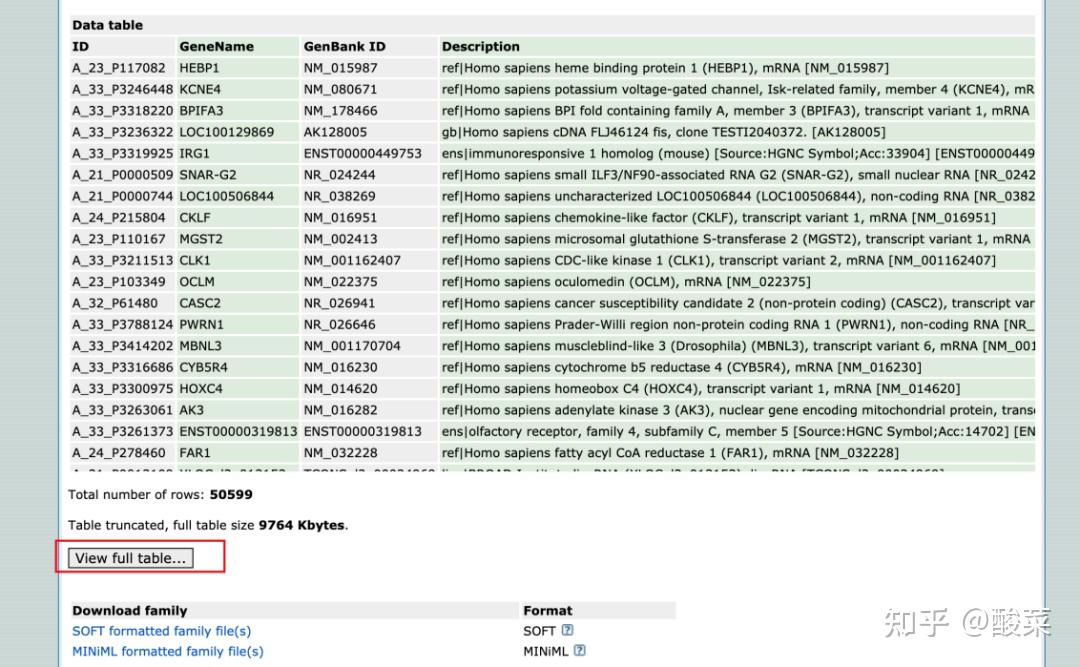
点击Full table下载 探针-分子 对应文件(如果是网页打开,可以在网页右键保存为txt,再用excel打开)。(注意:不是所有的平台都有具体的分子,有一些平台注释不全的,有可能找不到有注释分子名的,这种平台就不建议使用了)
如果Full table没有办法打开,则可以下载下方的soft文件,一样可以用excel打开(如果没有办法解压,说明文件没有下载全,需要重新下载。一般能完整下载的就能打开。):
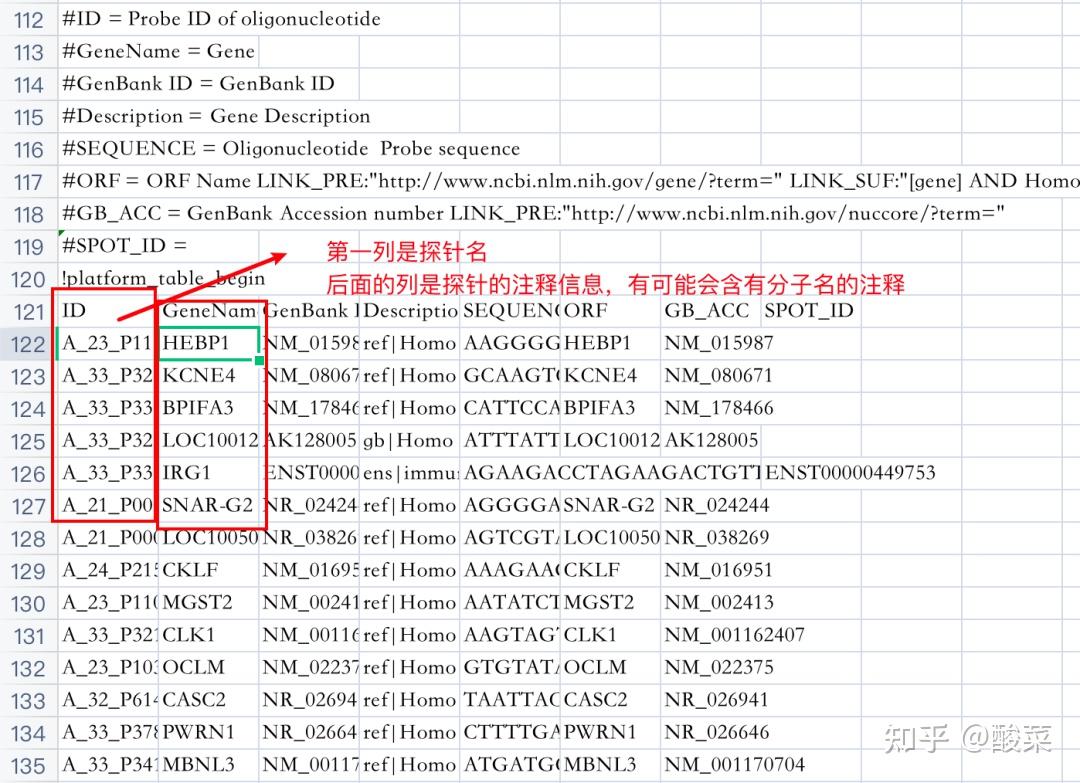
在GPL文件(探针-分子)中,先找到分子对应的探针号。
(如果一个分子对应多个探针,可以只挑其中一个或者挑“结果比较好”的探针)
(如果一个分子对应的探针对应了多个分子,这种探针也是可以用的,这种探针只是刚好检测的是他们共有的部分)
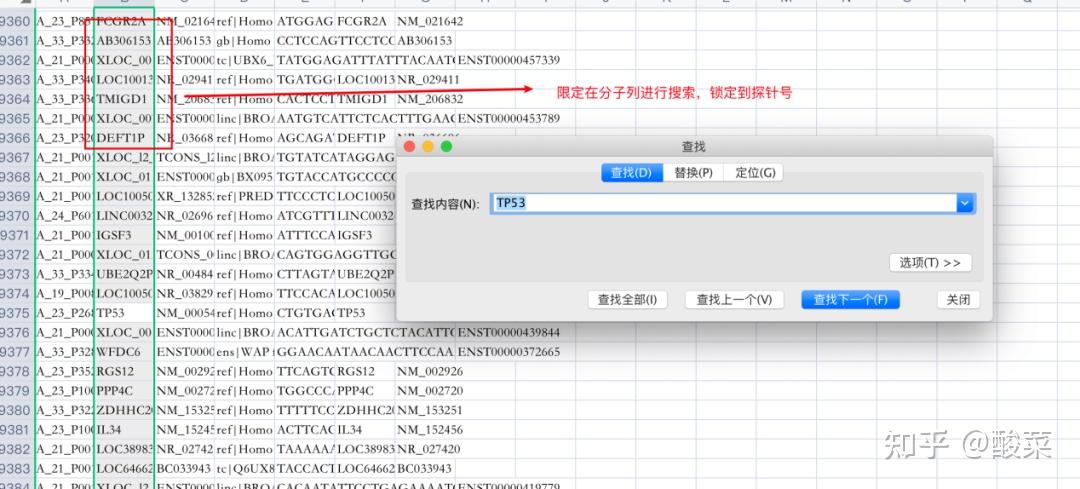
确定了探针后,在第一个下载的文件series_matrix文件中搜索这个探针号,找到对应的表达数据:
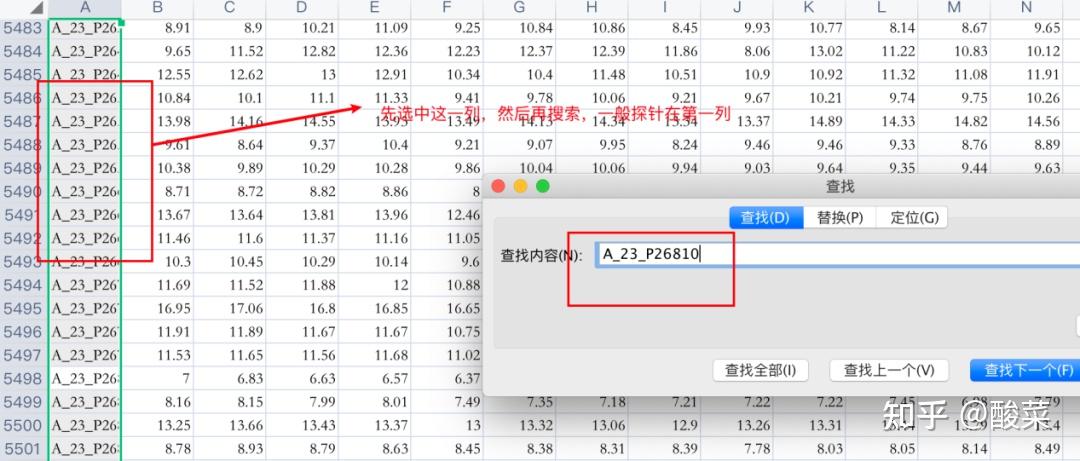
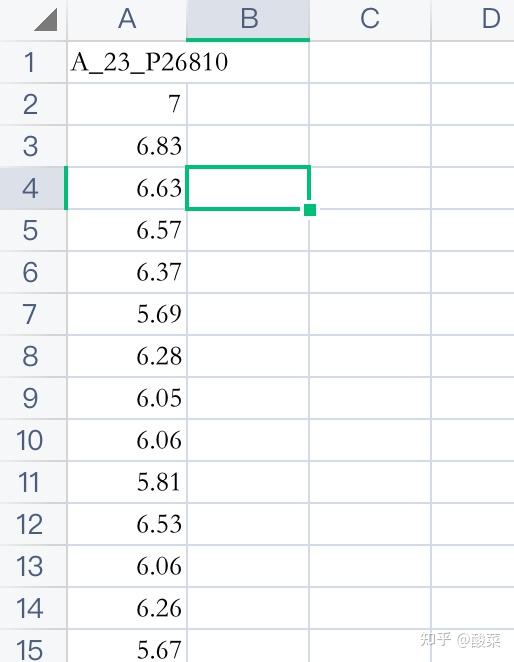
提取这个数据到一个新的excel表格中:
(复制-黏贴过程可以选择 转置)
同时,再回到series_matrix文件中提取样本编号和对应的临床信息到上面提取了分子表达的表内:

同样使用黏贴-转置的功能,再适当删除一些没用的列:
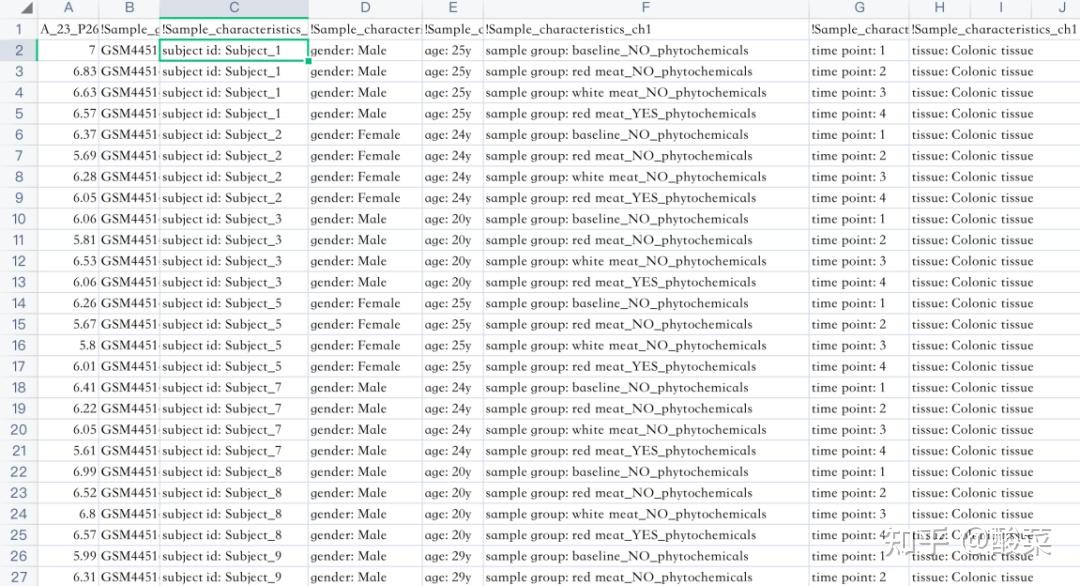
这样子我们就提取好了基本需要的信息(分子和对应的临床信息),可以用这个表格再根据不同的上传数据的模块进行数据格式的整理和上传了。
模块:分组比较图(表达差异)
简单从上面整理好的数据提取需要的数据到一个新的表格(格式可以查看“教程文档”的数据格式说明部分内容),然后上传:

简单整理后上传,然后就可以进行分析和可视化

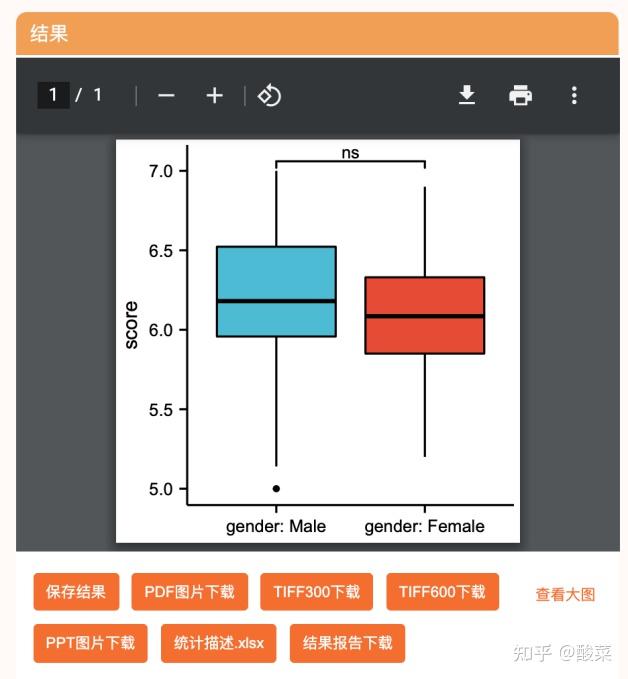
模块:诊断性ROC曲线
一样也是从上面整理好的数据中提取数据到一个新的表格中(格式可以查看“教程文档”的数据格式说明部分内容),然后上传:

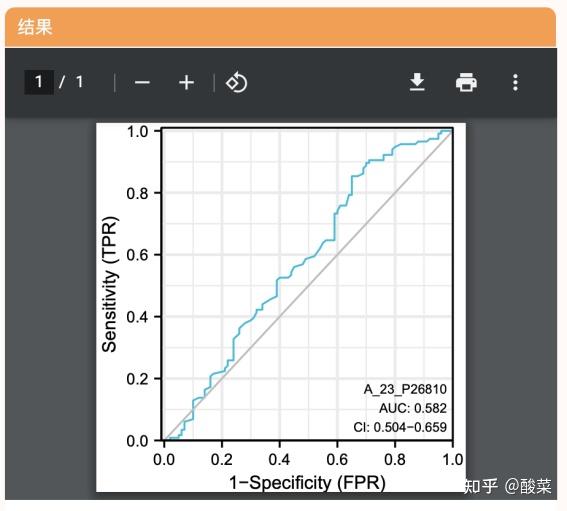
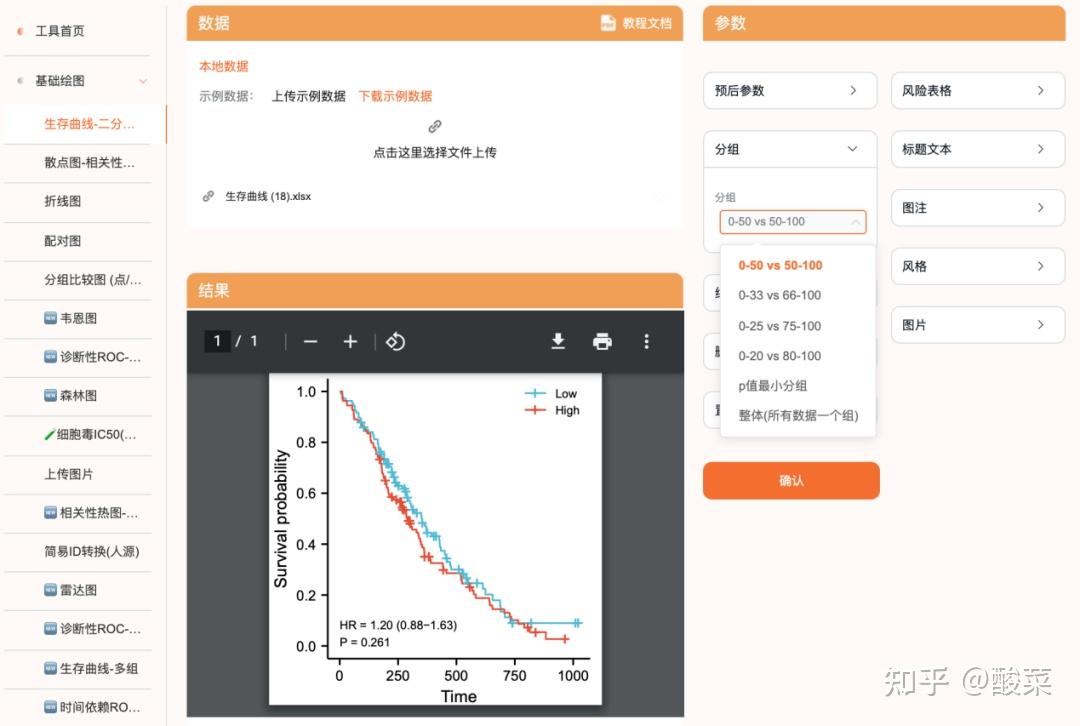
模块:生存曲线
当如果对应的数据集中提供了预后信息的数据(也是在series_matrix文件中的临床信息部分可以提取)(有预后数据也都是可以从series_matrixe文件中去的,不是所有的数据集都有预后数据,如果没有提供预后数据那么就是没有),则可以分析某个分子的预后KM图:
(这里是用了模拟生成的假的预后数据,只是为了演示用)
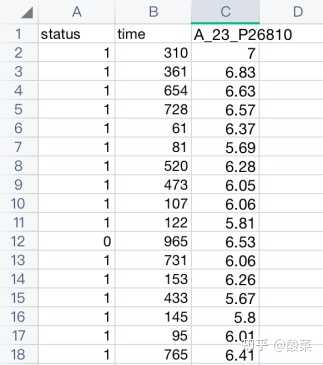
模块:其他上传模块
同样也是从上面整理好的数据提取需要的数据到一个新的表格(格式可以查看“教程文档”的数据格式说明部分内容),然后上传数据进行分析。
酸菜:史上最强GEO一站式整理分析教程!独家诀窍!建议收藏!
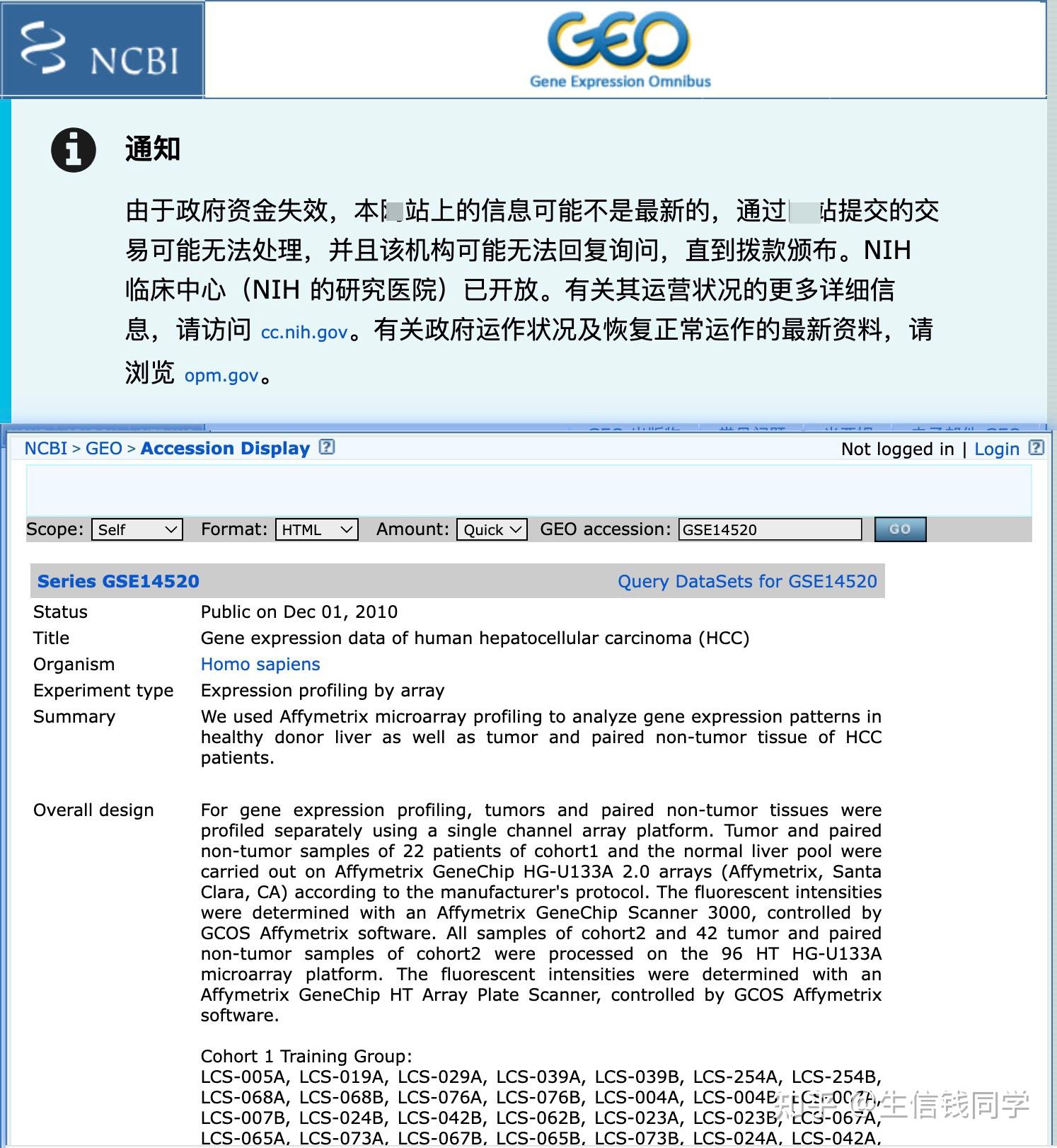
天塌了,GEO的生信数据其实开源性做的非常好,基本上数据都能下载,很多纯生信的文章都是靠挖的这个数据。现在进入任意一个数据中都能看到这个通知,不再更新了,看看未来 到了,还能不能恢复;
版权声明:本文内容由互联网用户自发贡献,该文观点仅代表作者本人。本站仅提供信息存储空间服务,不拥有所有权,不承担相关法律责任。如发现本站有涉嫌侵权/违法违规的内容,请联系我们,一经查实,本站将立刻删除。
如需转载请保留出处:https://51itzy.com/kjqy/244942.html