
今天带大家学习一款基于GEO数据库的基因表达分析及可视化功能数据库,GEOexplorer,顾名思义,帮你解决那些年因学R偷的懒而错过的美好数据集,一起来看看吧~!

GEOexplorer数据库(https://geoexplorer.rosalind.kcl.ac.uk/)于2022年5月份发布在Nucleic Acids Research杂志,基于GEO数据库芯片和RNA-seq数据或者上传个人数据集,提供探索性数据集分析、基因表达差异分析和功能注释及可视化等功能,产生丰富的交互式可视化结果,无需要编程即可产出发表级图表,为生命领域研究人员尤其是缺乏生物信息学技能的研究者高效利用公共数据集挖掘重要信息提供极大便利。

菜单栏Tutorial部分提供数据库介绍及详细的数据库使用指南。其中Introduction部分可见数据库提供以下功能:数据集检索、数据集探索性分析、多个数据集整合及批间差校正、基因表达差异分析、差异基因功能富集和可视化等。除在线网站之外,用户还可以选择安装GEOexplorer R包进行基因表达差异及功能注释相关分析。

1、GEO Search功能
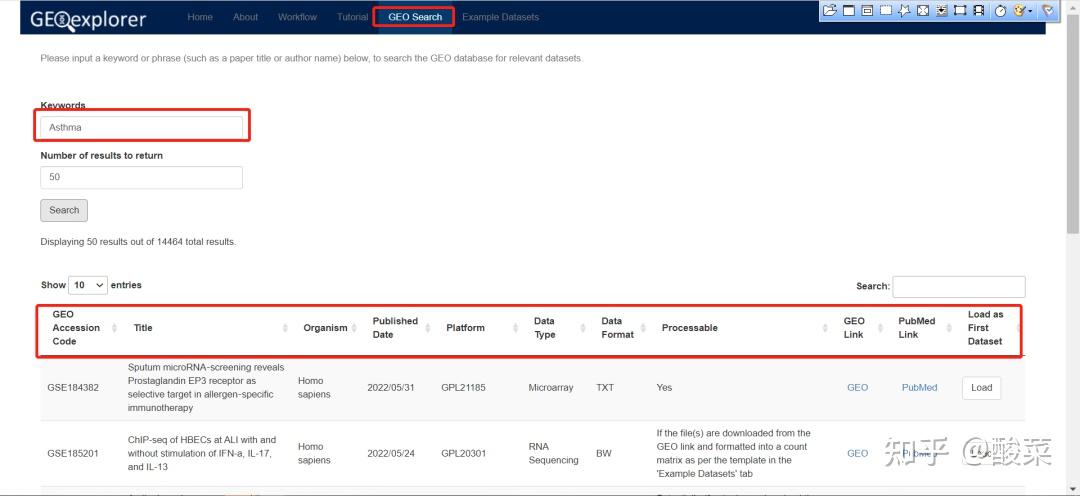
GEO Search功能模块支持以关键词检索感兴趣的数据集或样本,以哮喘(Asthma)为例,结果显示相关数据集或样本有14464个,结果界面不大方便进行数据集筛选,因此这个功能略显鸡肋,大家仍旧可以在GEO检索到感兴趣的数据集之后,再来借助GEOexplorer数据库进行后续系列分析。
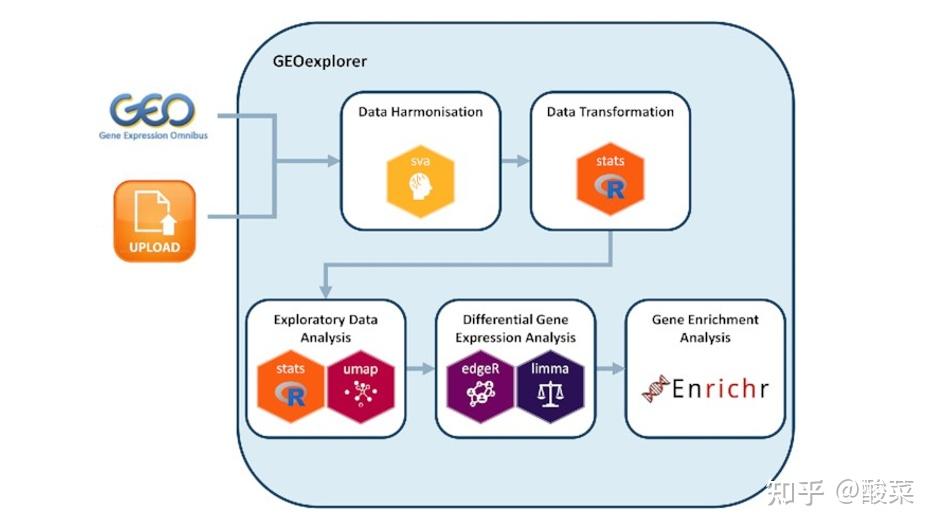
2、数据分析功能
借助GEOexplorer数据库进行数据分析有四步:(1)Dataset Information,查看数据集基本信息;(2)Exploratory Data Analysis,探索性数据分析;(3)Differential Gene Expression Analysis,基因表达差异分析;(4)Gene Enrichment Analysis,基因富集分析。
这里跳过GEO检索数据集过程,新手小伙伴感兴趣的话可以学习单元课《GEO数据集使用教程》,或参加GEO数据库挖掘成长营。以PMID: 文献提供的哮喘数据集GSE4302, GSE43696, GSE63142, GSE67472和GSE41861为例。
单个数据集分析
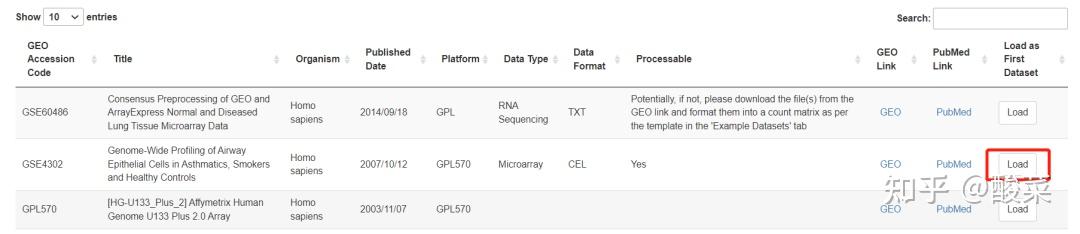
首先,检索并加载目标数据集。在GEO Search检索目标数据集并点击load加载,或直接在数据分析功能页面选择单个数据集、选择芯片或测序数据、输入目标数据集及平台号,以GSE4302为例,点击Analyse提交分析任务。注意如果是RNA-seq数据需要判断是否需要log转换,简单的办法就是看基因表达谱是否有小数,如果有则已经过log转化,则不建议用GEOexlporer继续分析。
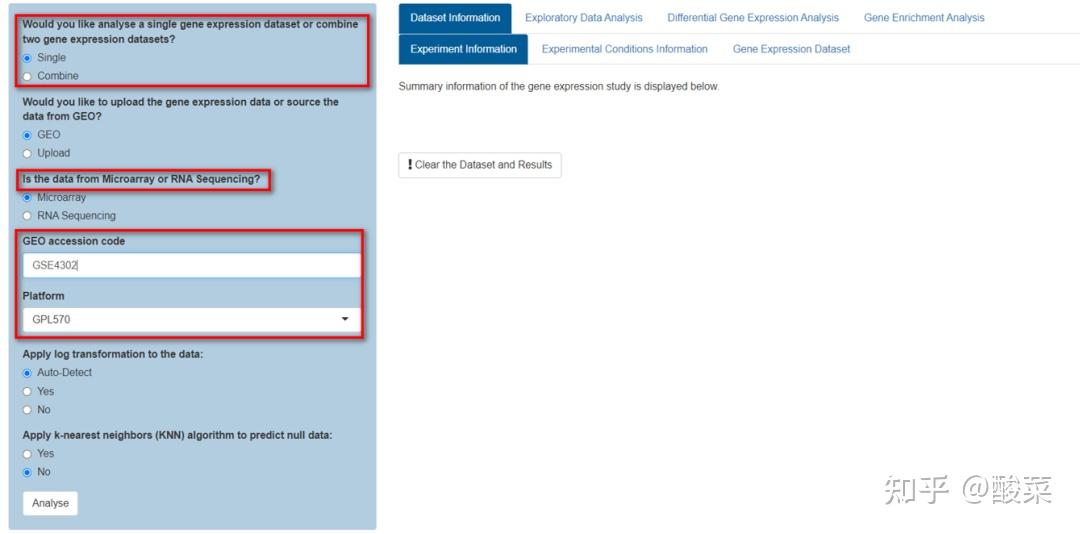
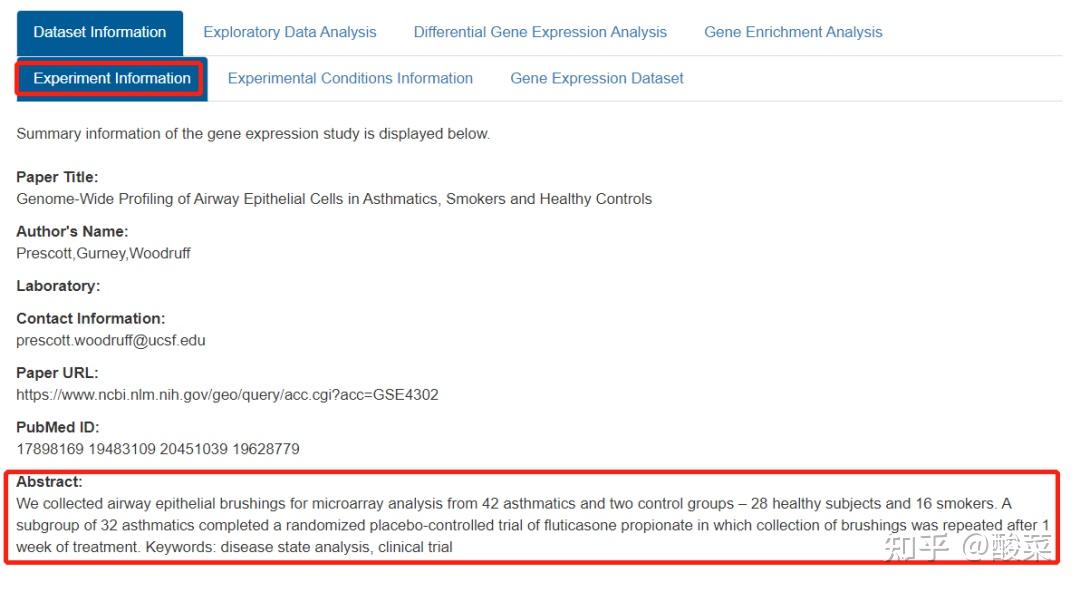
(1)Dataset Information:数据集基本信息
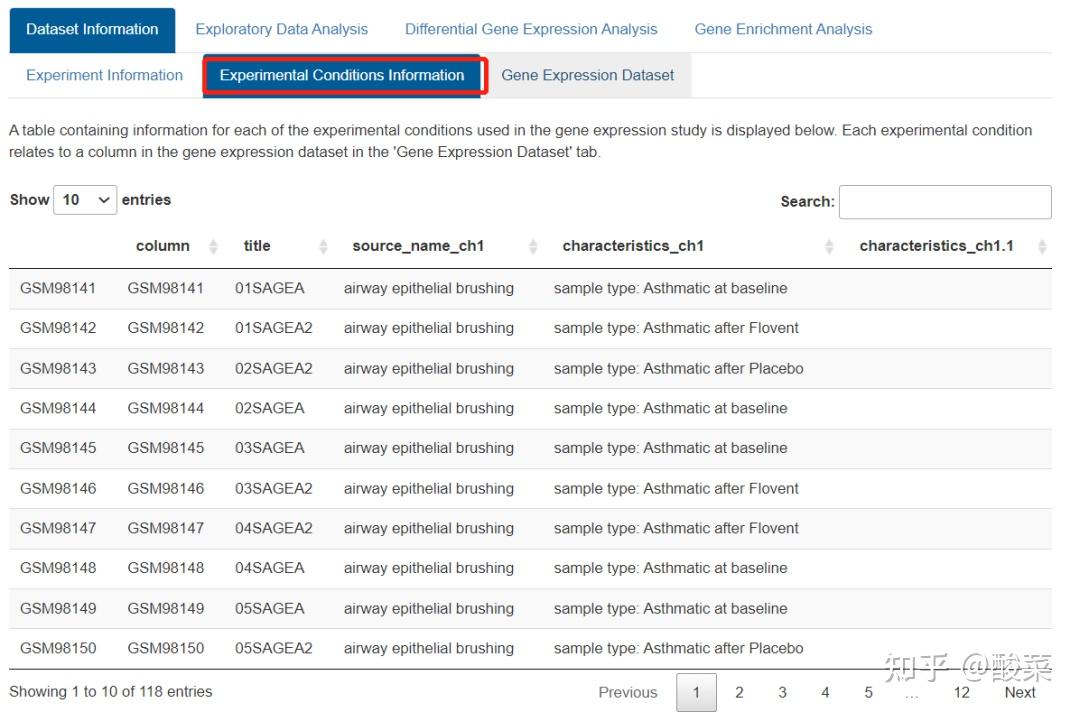
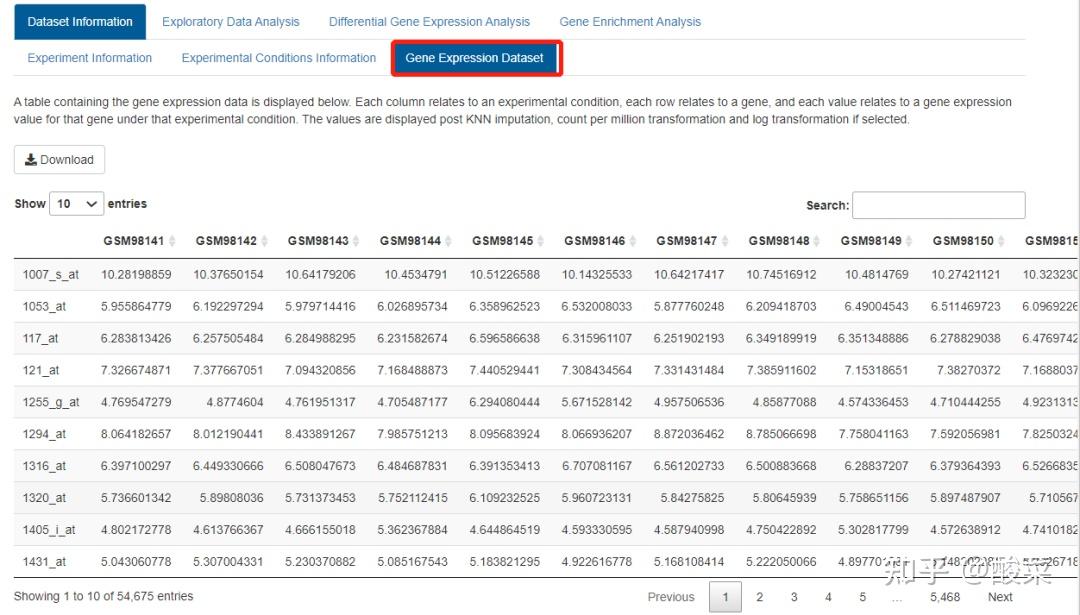
Experiment Information提供GSE4302数据集基本信息,我们看一下Abstract可以了解作者做了什么工作;Experimental Conditions Information提供样本及其分组信息;Gene Expression Dataset为基因表达谱。
(2)Exploratory Data Analysis:探索性数据分析
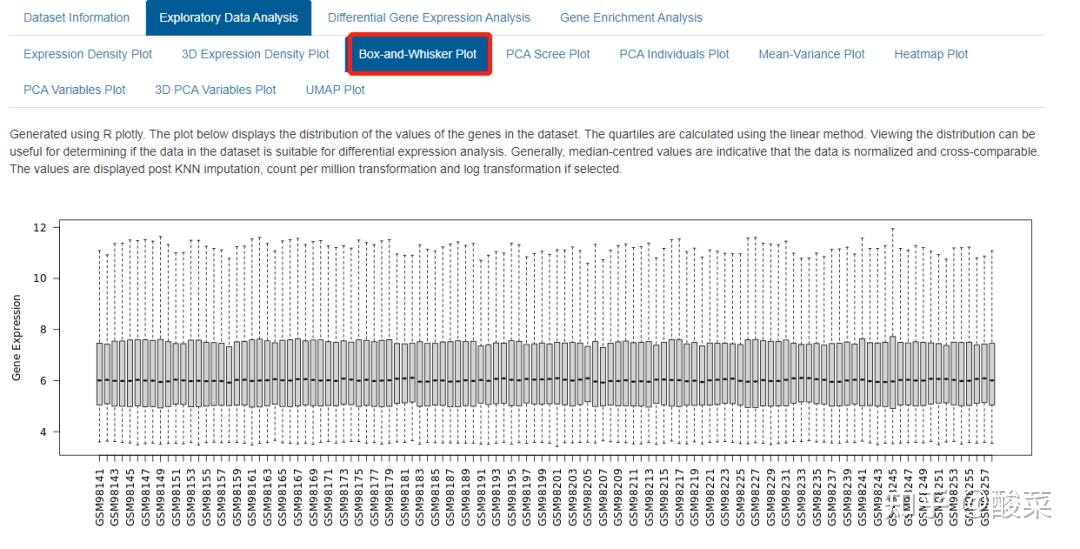
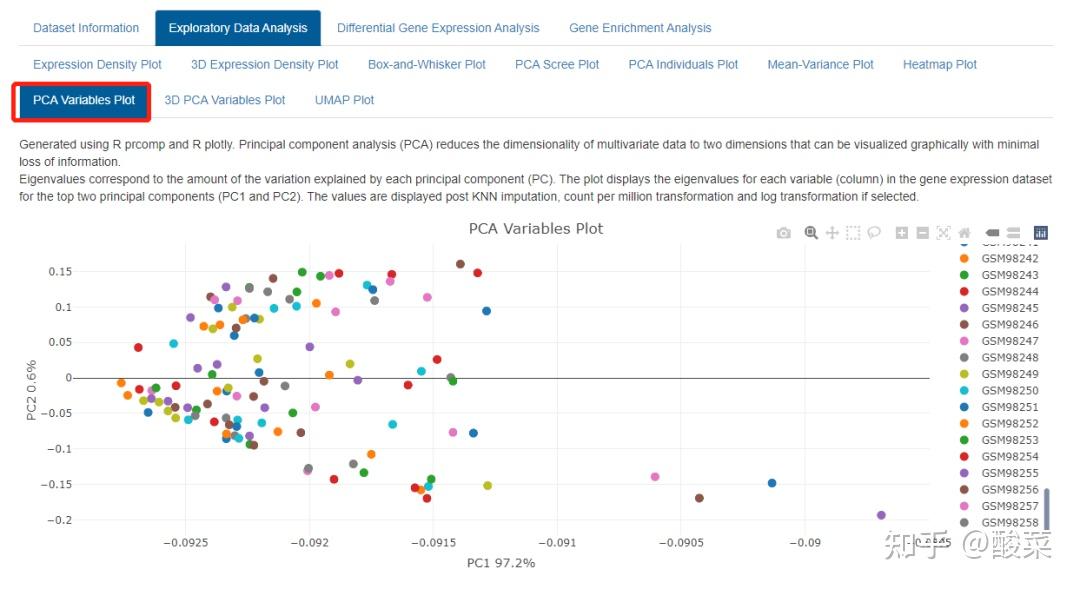
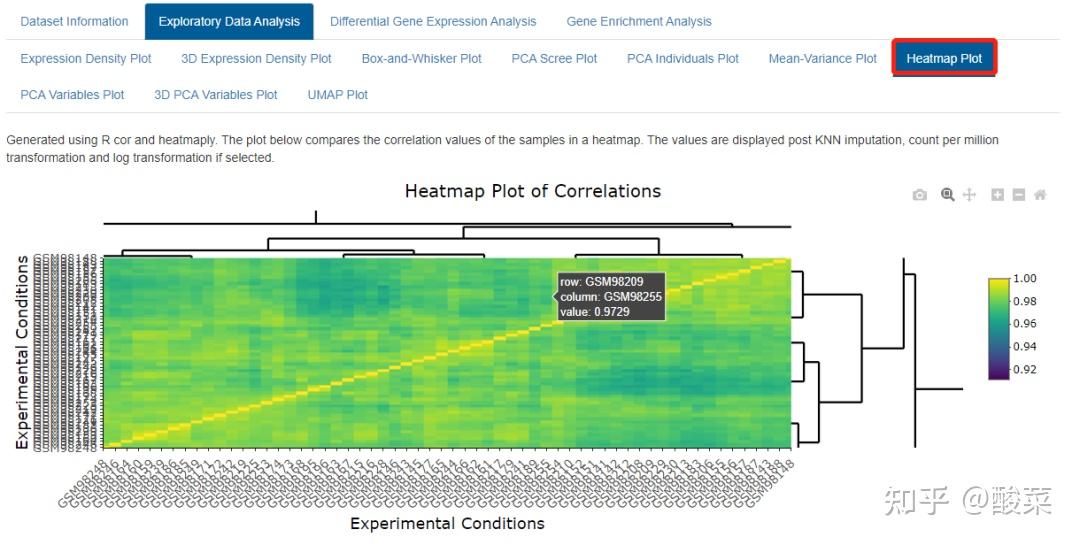
事实上就是数据预处理和质控环节,重点关注Box-and-Whisker Plot、PCA Variables Plot和Heatmap Plot图,查看数据质量。箱图展示各样本均数不在同一水平,大概率是数据集尚未进行标准化处理;若PCA图分的比较开、热图按组别聚类,基本上数据质量可以,而本例样本太多导致PCA和热图很难辨别聚类情况。
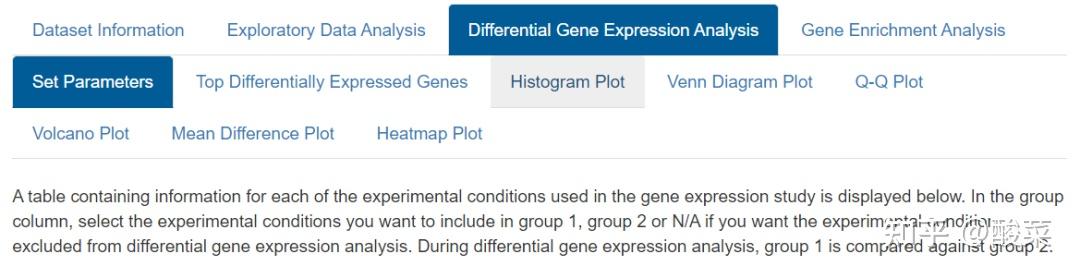
(3)Differential Gene Expression Analysis:基因表达差异分析
点击Set Parameters设置参数:选择Group1与Group2纳入的样本进行比较,以基线哮喘和健康对照为例;再选择P值校正方法、是否强制标准化、是否应用limma vooma,及显著性水平截断值。在Tutorial部分有说明:对原始RNA-seq数据需要强制标准化和limma vooma处理,对芯片数据如果在前述箱图展示样本均线不齐、组间差异比较大或是密度图密度曲线不呈正态分布时,建议强制标准化和进行limma vooma处理。
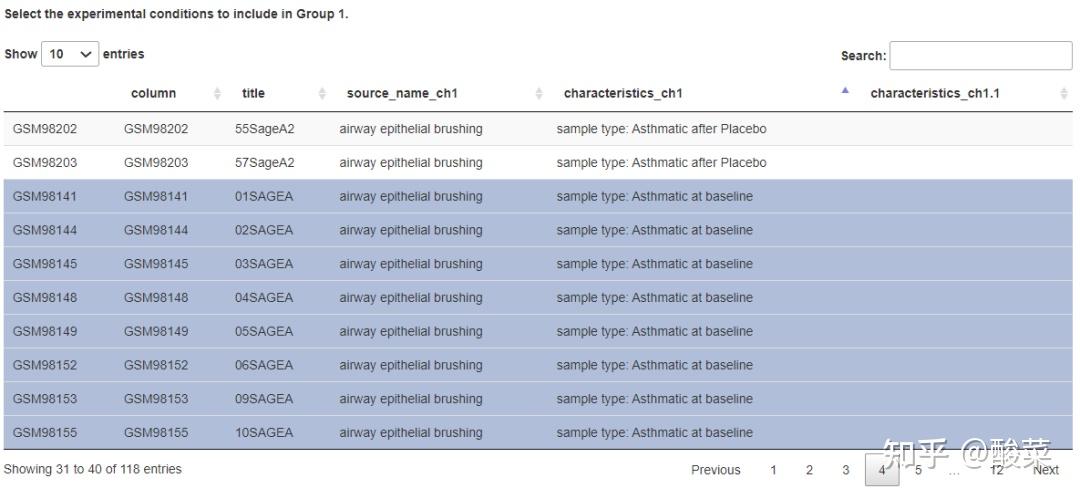
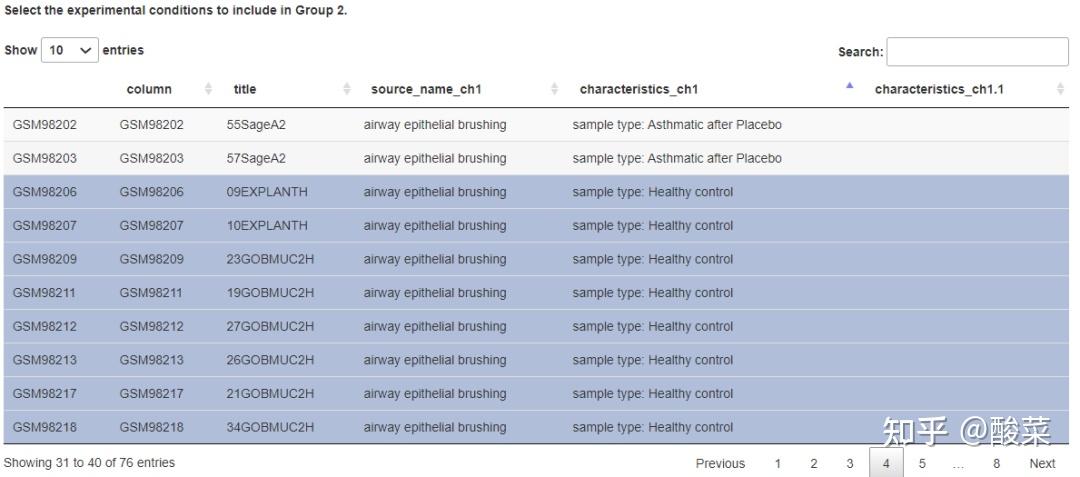
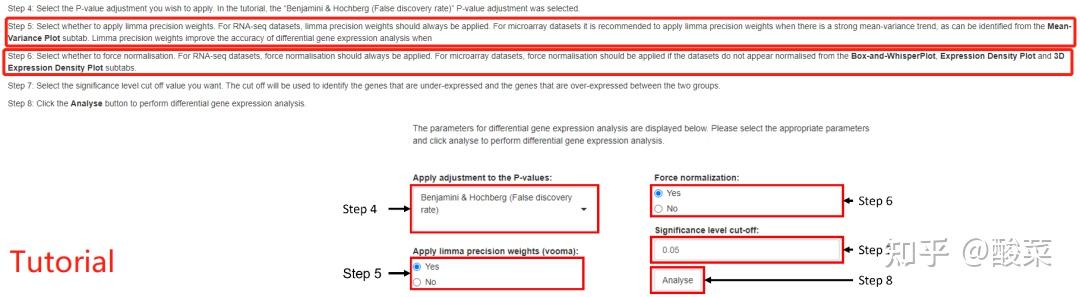
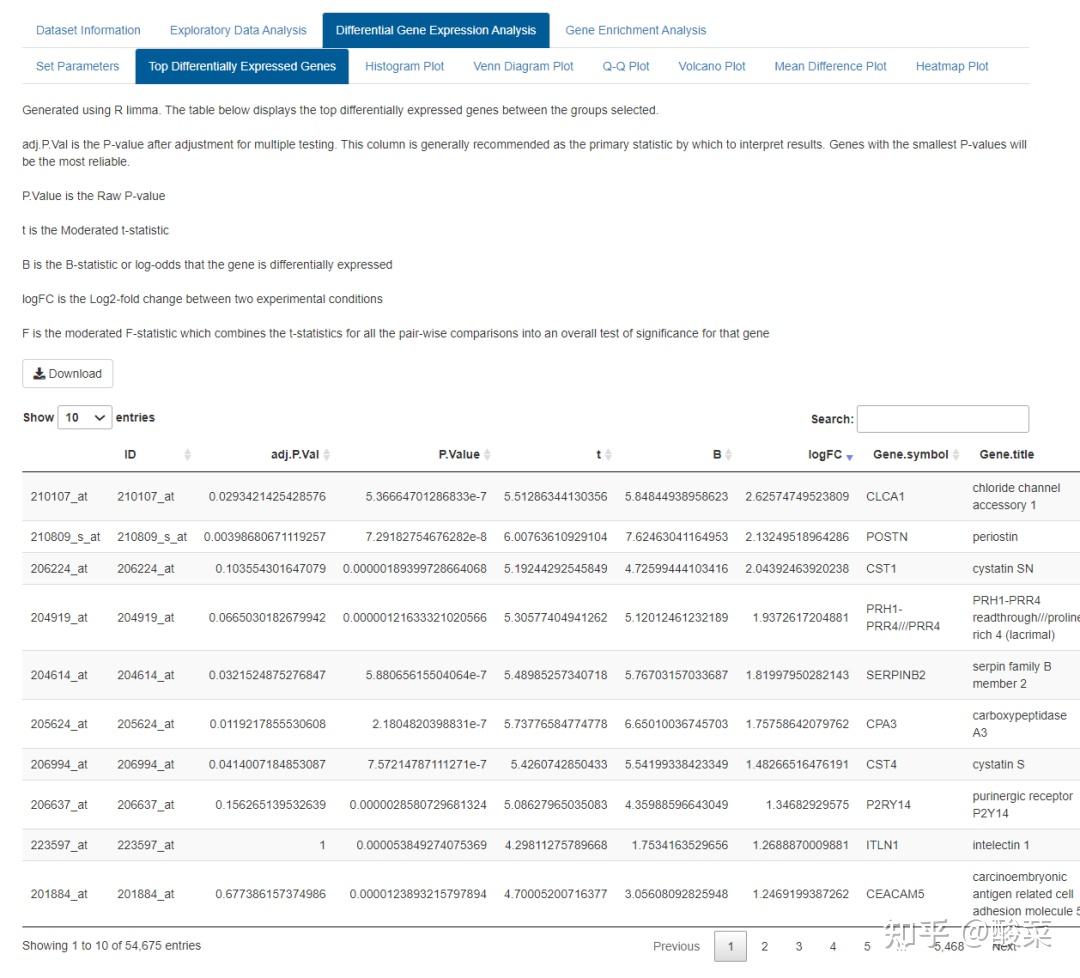
点击Analyse获得表达差异分析结果:Top Differentially Expressed Genes表格展示Top表达差异基因信息;提供表达差异基因Volcano Plot和Heatmap Plot。
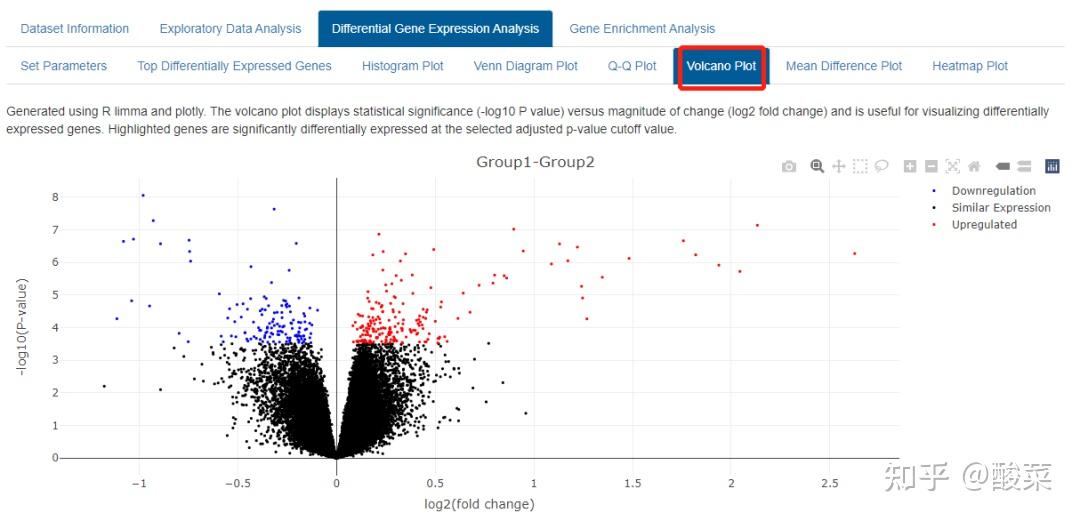
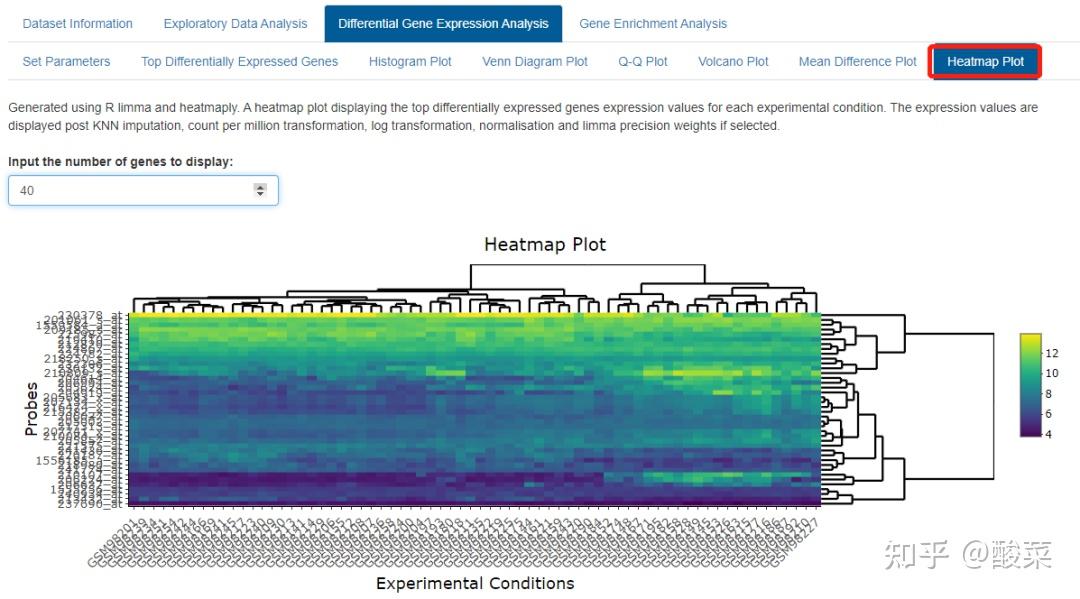
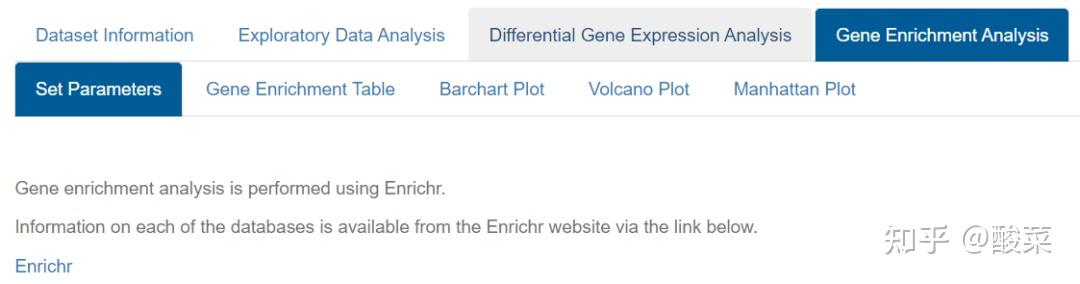
(4)Gene Enrichment Analysis表达差异基因富集分析
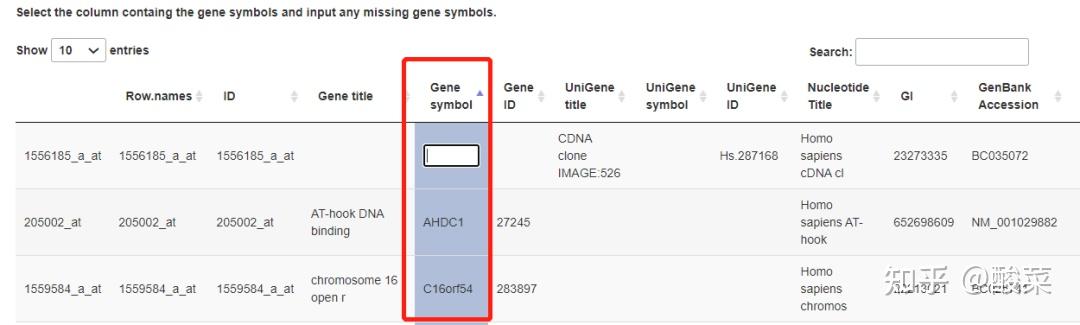
点击Set Parameters设置参数:选择Gene symbol列,双击空白处手动填补;选择需要分析的数据库。点击Ananlyse获得分析结果。
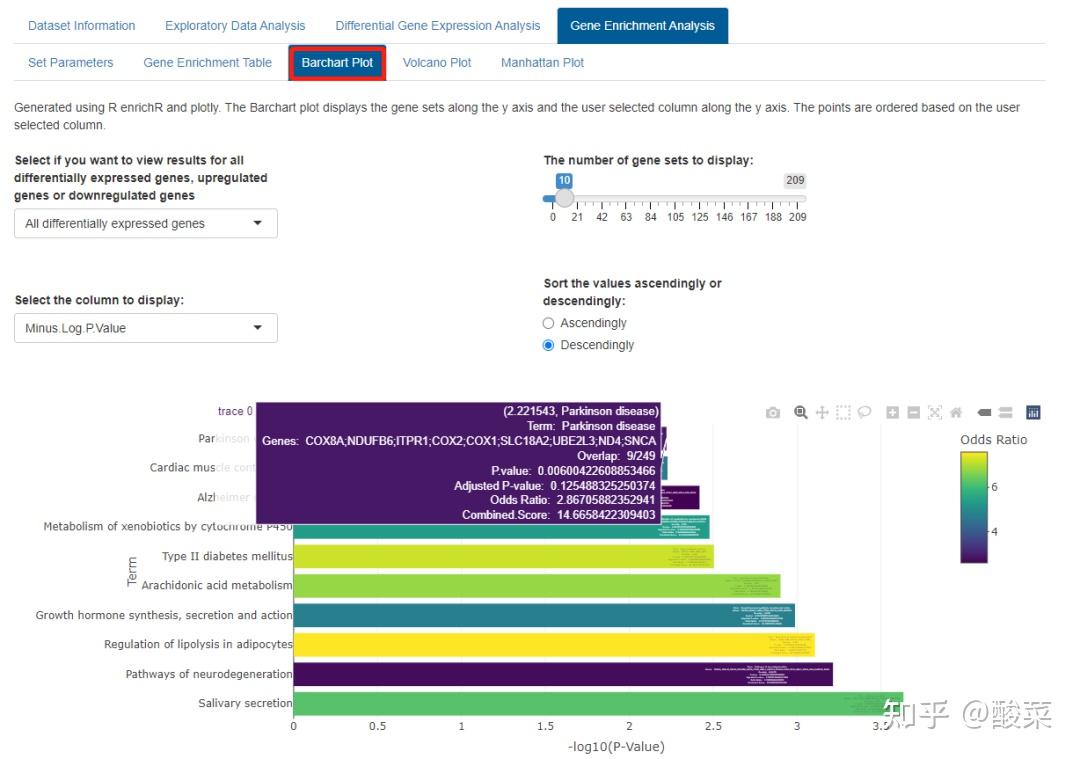
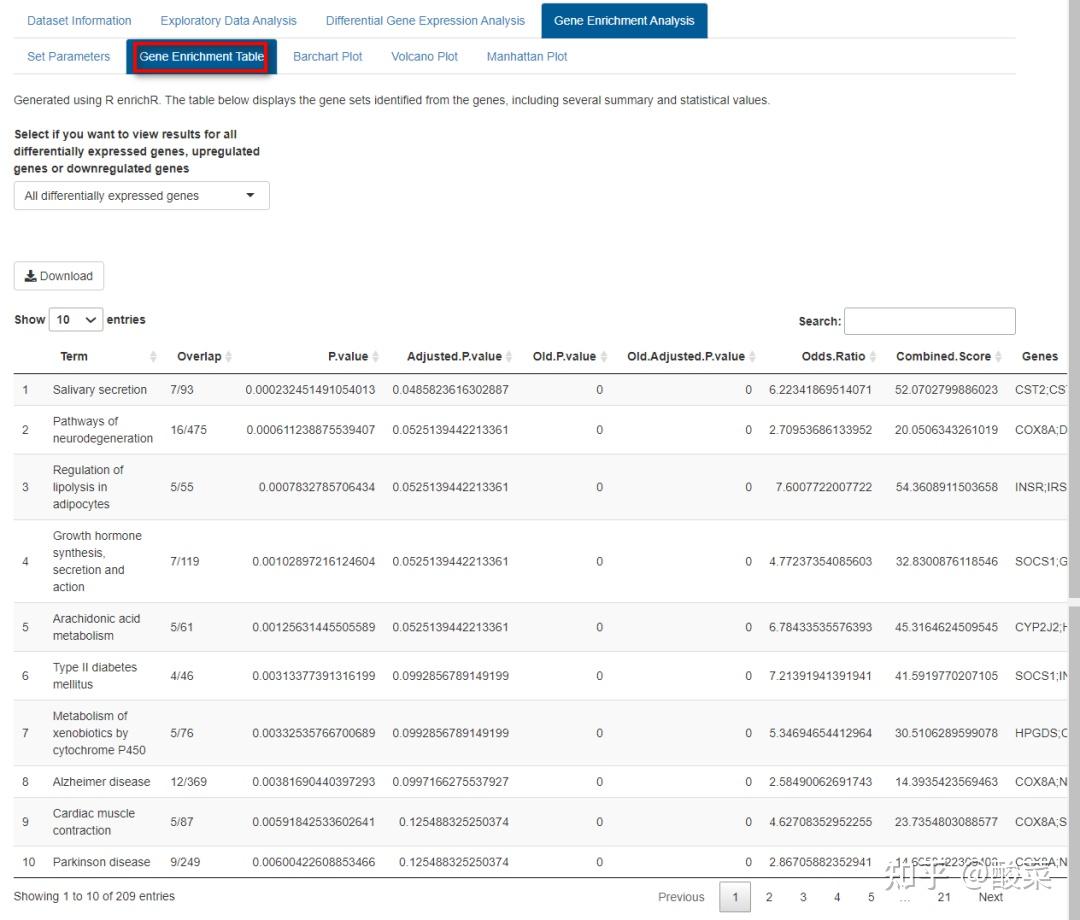
查看富集分析结果,以表格和柱状图形式展示,鼠标悬停在柱子上可以查看具体基因、通路信息和P值。可以选择上调、下调或全部基因。
两个数据集分析
同样的方法,检索并加载目标数据集。直接在数据分析功能页面选择多个数据集、选择芯片或测序数据、输入目标数据集及平台号,以GSE4302和GSE4302,其他参数默认,批间差校正的方法选择贝叶斯,点击Analyse。结果基本类似。
重点来看Exploratory Data Analysis,其中Expression Density Plot图、Box-and-Whisker Plot图、聚类热图和PCA图均显示出明显批次效应,即样本是按照批次进行聚类的而不是按照实验分组来聚类。因此上一步选择贝叶斯方法进行批次效应校正是有必要的。
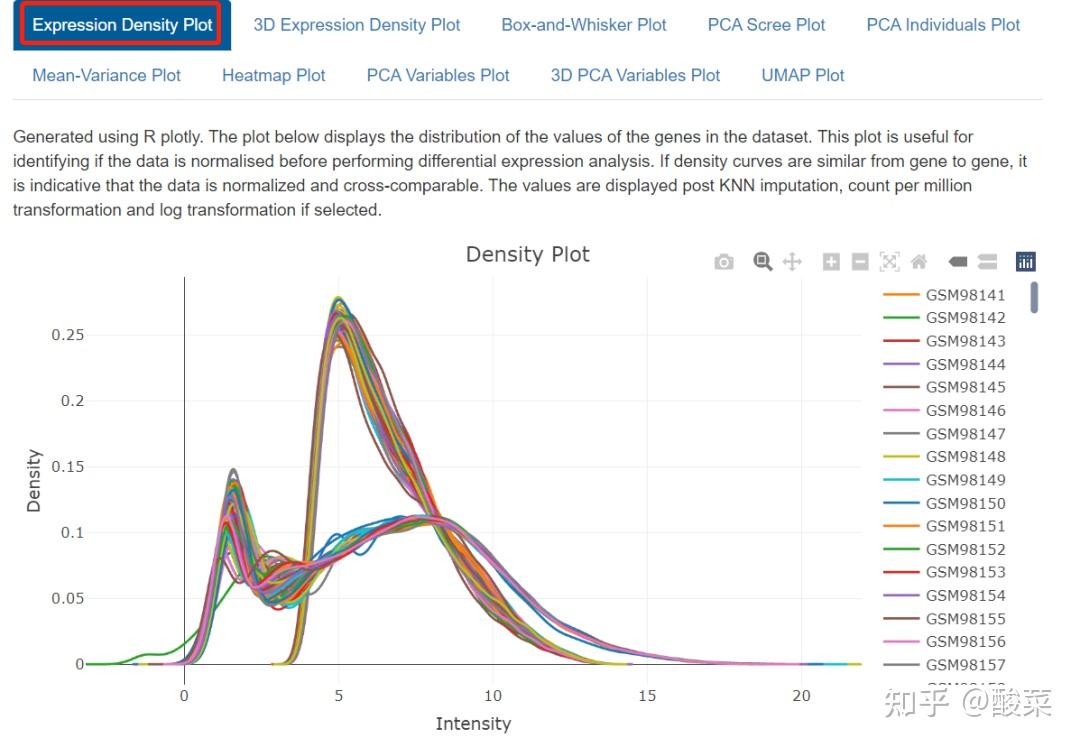
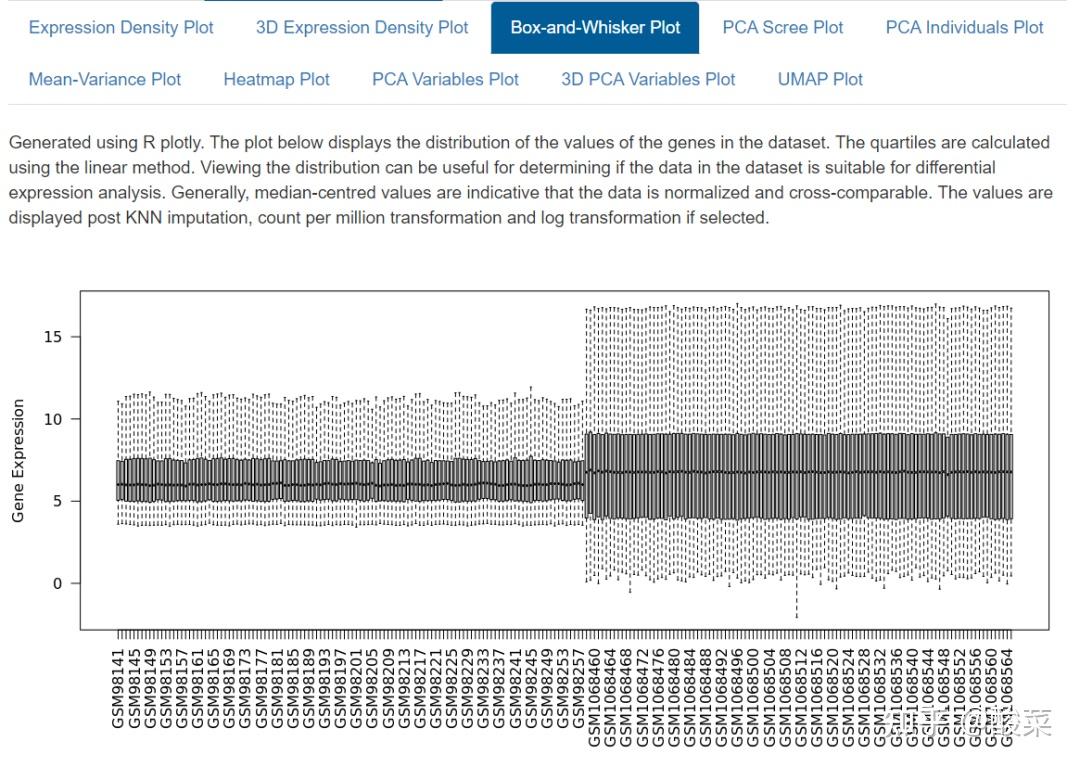
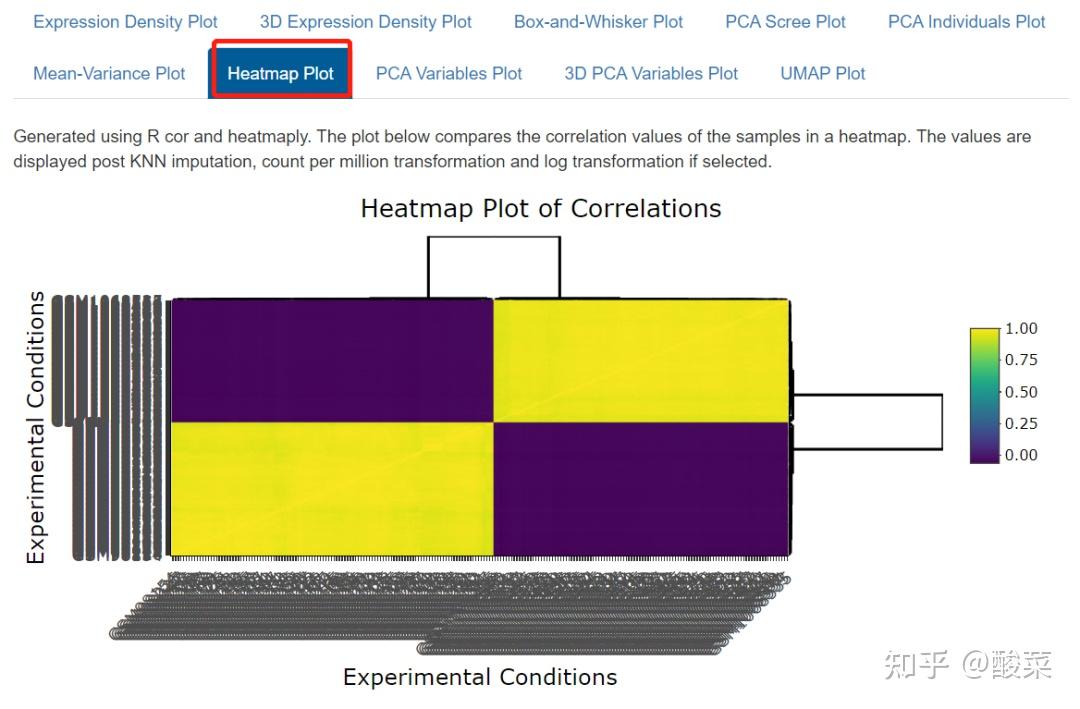
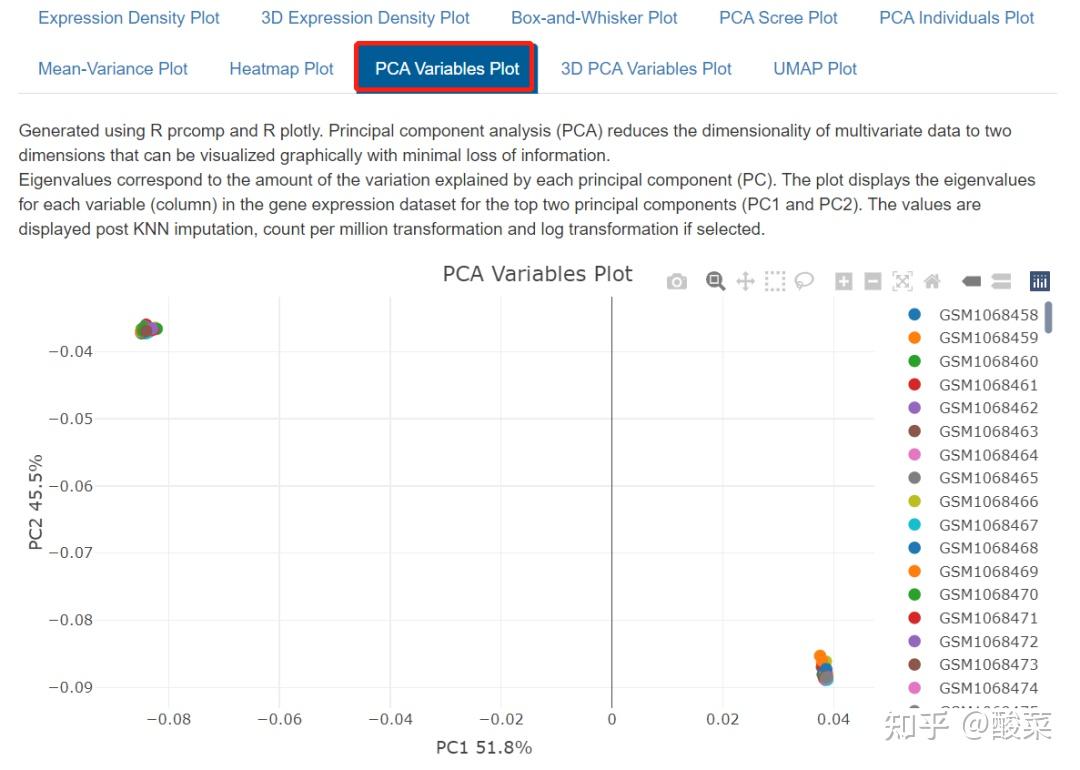
接下来的Differential Gene Expression Analysis和Gene Enrichment Analysis分析基本与前述类似,不再赘述。
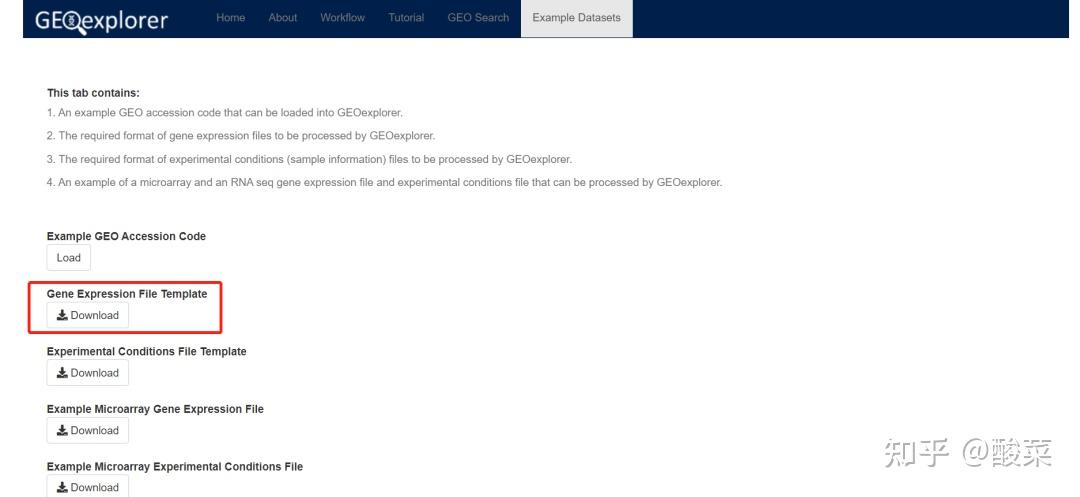
3、上传数据准备
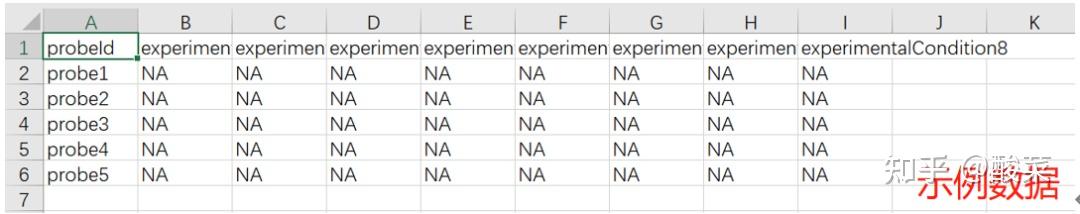
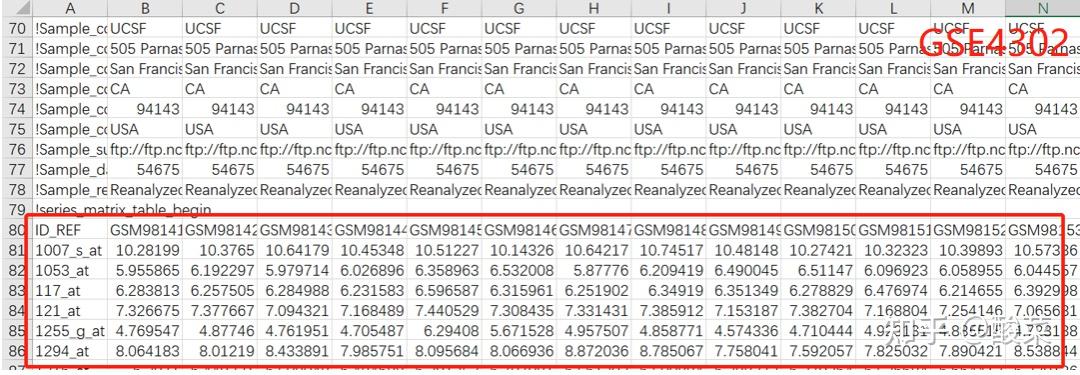
有些GEO数据集无法检索加载到数据分析功能模块,需要手动下载和整理。菜单栏处点击Example Datasets下载示例基因表达谱文件,Excel打开,即基因探针列+样本列,打开GSE4302表达谱,复制红框部分数据到示例文件中即可。
总结
GEOexplorer提供基于GEO数据集或上传个人数据集的探索性数据分析、基因表达差异分析和功能注释及可视化等功能,产生丰富的交互式可视化结果,无需要编程即可产出发表级图表,为生命领域研究人员尤其是缺乏生物信息学技能的研究者高效利用公告数据集挖掘重要信息提供极大便利。
以上就是GEOexplorer数据库全部内容,开发并维护数据库不易,小伙伴们使用时别忘记引用以下文献哦~!
Hunt GP, Grassi L, Henkin R, Smeraldi F, Spargo TP, Kabiljo R, Koks S, Ibrahim Z, Dobson RJB, Al-Chalabi A, Barnes MR, Iacoangeli A. GEOexplorer: a webserver for gene expression analysis and visualisation. Nucleic Acids Res. 2022 May 24:gkac364. doi: 10.1093/nar/gkac364. Epub ahead of print. PMID: .

之前我们已经和大家分享过如何上传GEO数据,同样,我们也可以从公共数据库下载其他研究者的数据进行分析,探寻自己感兴趣的研究方向。这种通过对现有公共数据挖掘数据进行分析的研究方式目前正是生物信息分析的热门领域。GEO数据库储存着海量的二代测序数据(特别是转录数据)和芯片数据,就像一座“藏宝山”,我们可以通过挖掘前人研究数据,提取相关研究结果,构建自身研究方向的基础架构,再辅以“湿实验”验证或针对性的其他组学测序分析,最后解决自身研究方向的复杂问题。那么接下来小诺将为大家带来全套的GEO数据挖掘系列教程,包括五步:
搜寻数据集及下载数据 差异分析—>差异表达基因 —>五大数据库注释—>蛋白互作等网络和通路注释
诺禾致源:数据上传 | GEO数据上传操作指南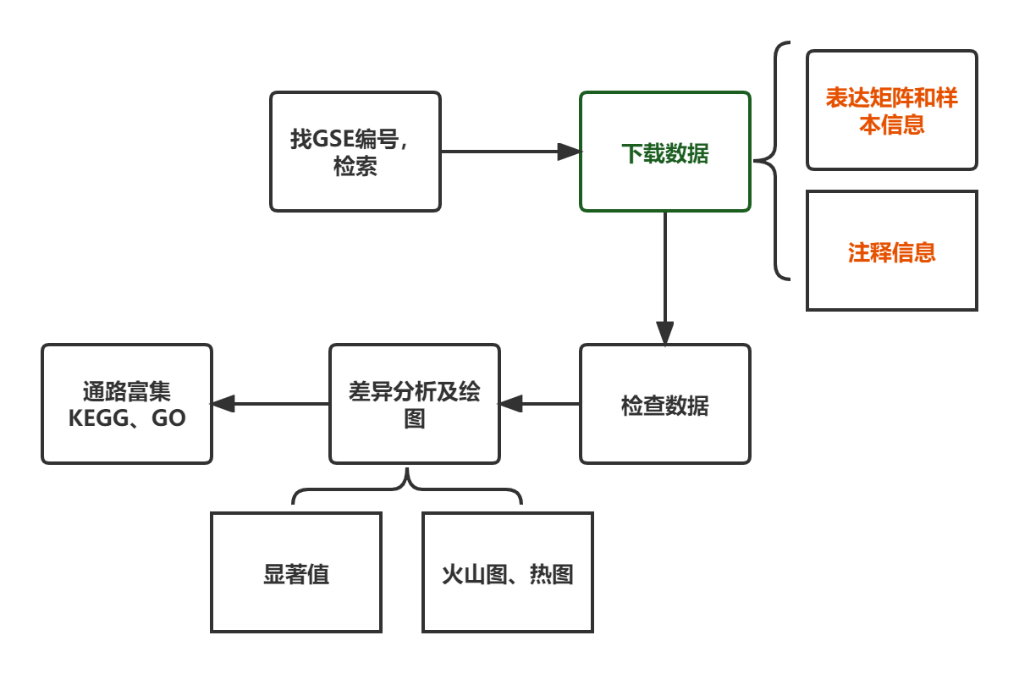
今天先为大家分享第一步“数据下载”,以便后续挖掘感兴趣的基因及构建通路。
首先我们可以需要先寻找贴合我们研究方向的论文所使用的数据集,下载里面的GPL文件和表达矩阵“series matrix”做基因的表达分析;再依据包含样本生存数据的临床特征数据集,去做生存性分析。
在进入GEO数据库官网前,需要先理解一下GEO的数据编号含义:
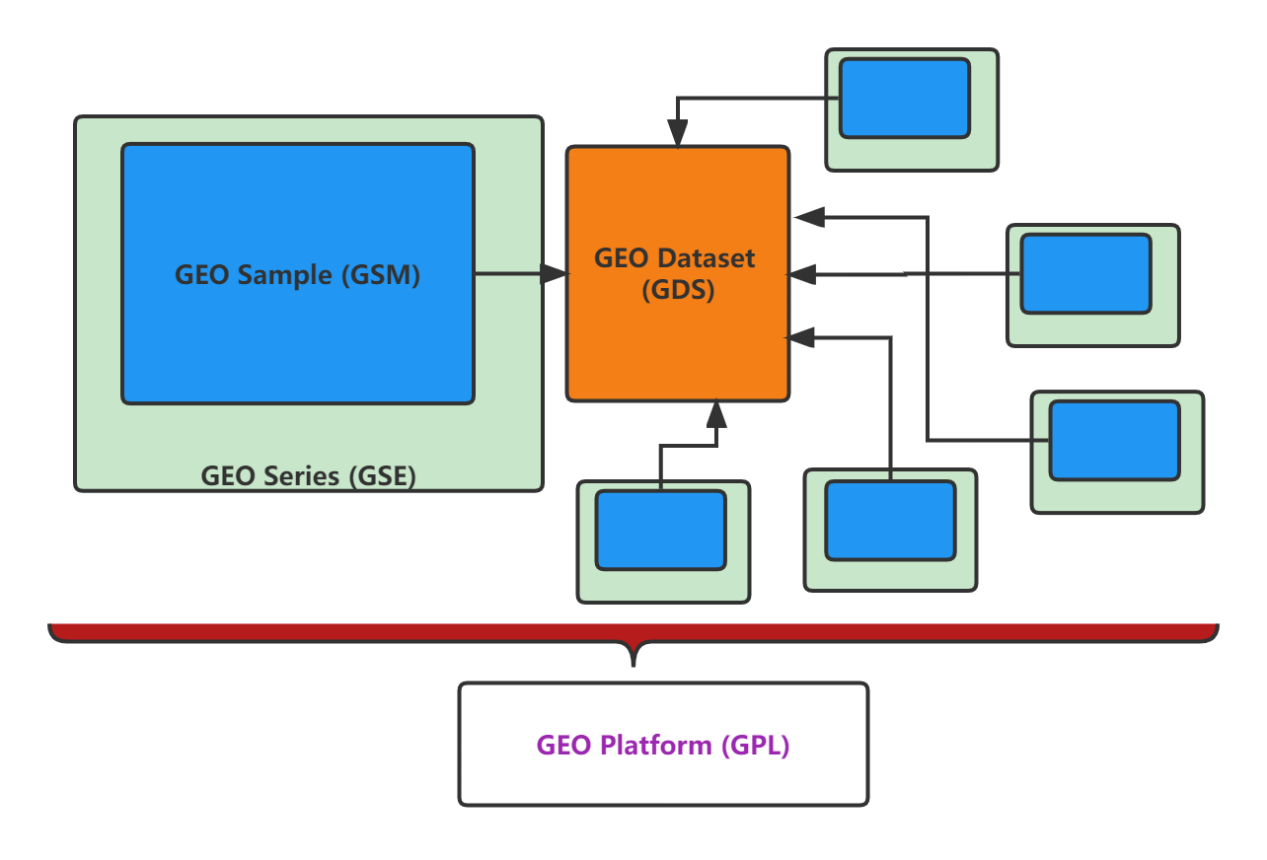
一篇文章可能包含至少一个GSE数据集,一个GSE数据集里面可能包含至少一个GSM样本。多个研究的GSM样本根据研究目的会整合为一个GDS,不过GDS运用的很少。而每个数据集都有着对应的芯片平台,就是GPL。
首先,登录GEO官网 https://www.ncbi.nlm.nih.gov/geo/,在右侧的搜索框输入一个GSE号,我们以gse21933为例,在搜索栏输入后点search进行检索。
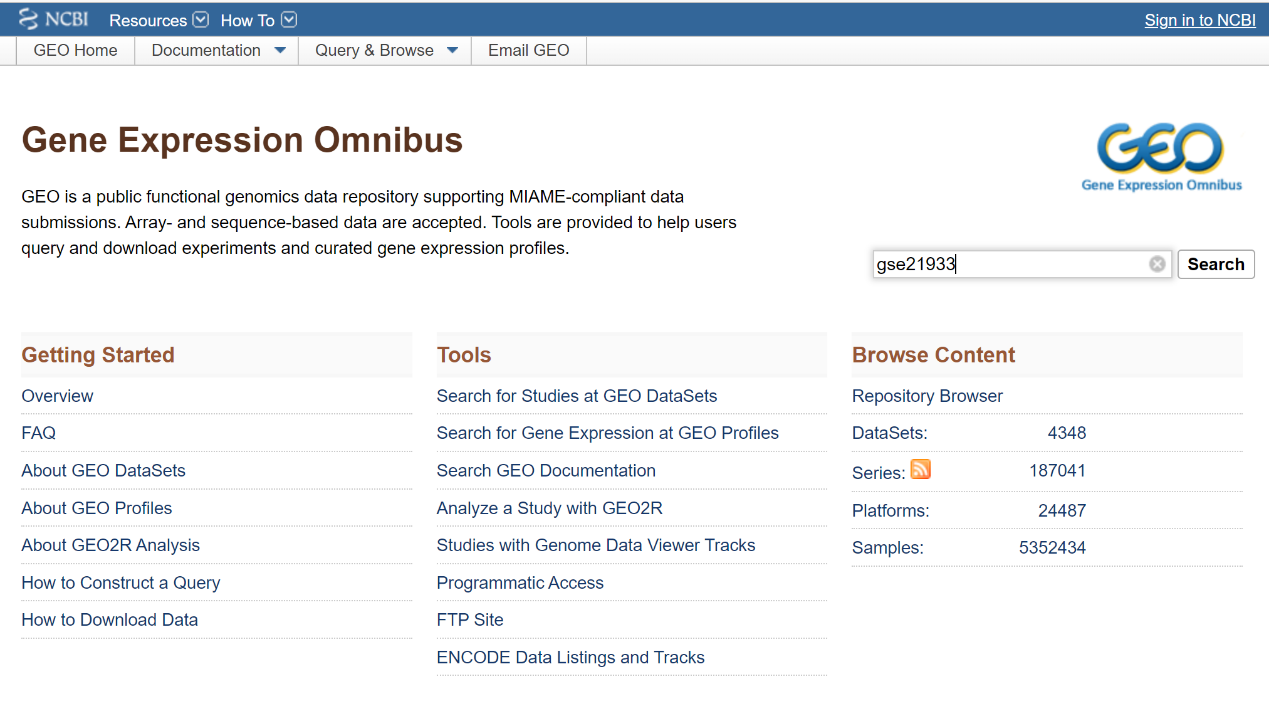
检索后会进入Accession Display界面,里面包含着这个GSE数据的基本信息,如:标题、物种、研究概要、作者、样本描述、测序平台等等,当然还有我们最需要的原始数据。
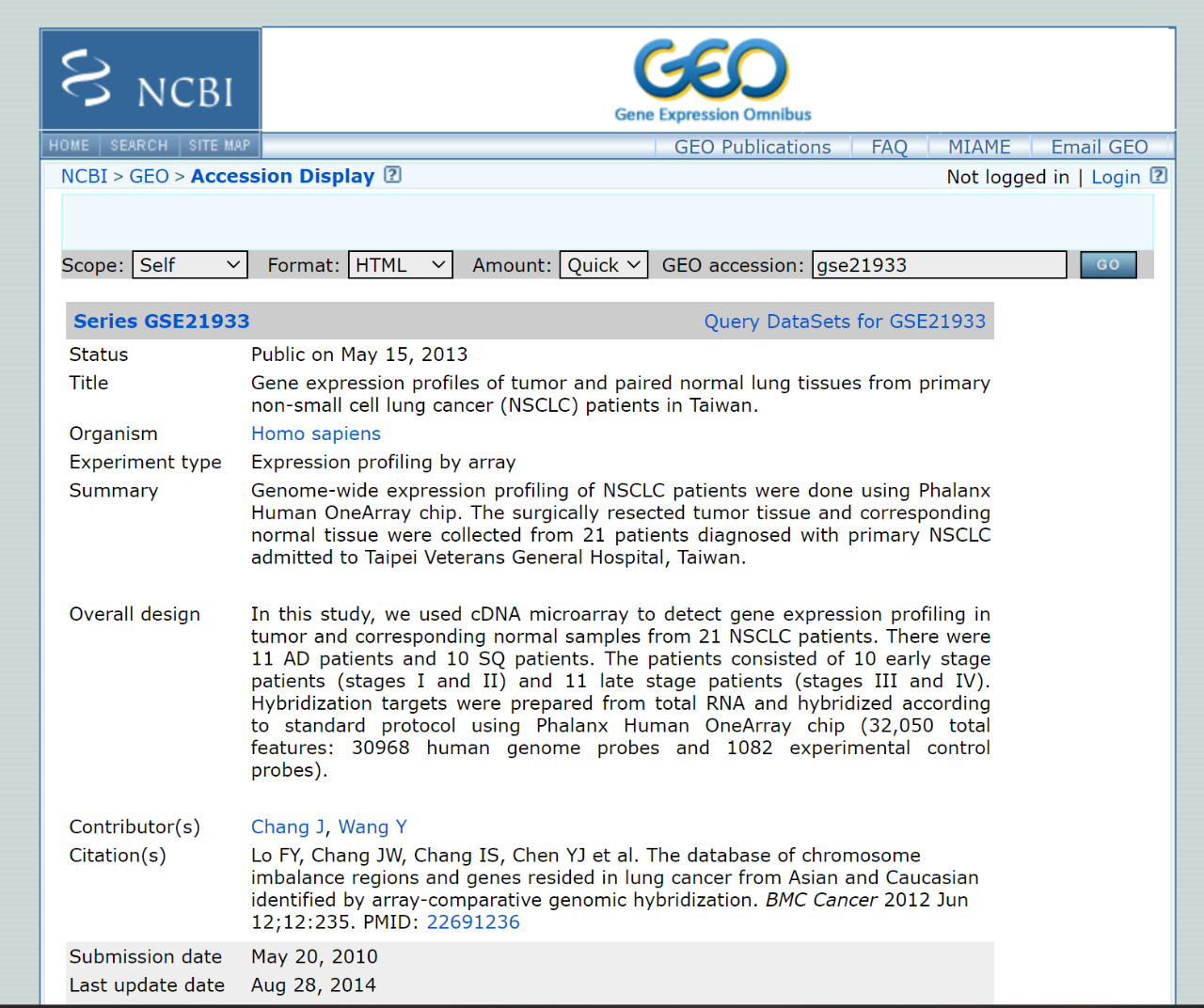
上图可知道,这组数据包含的是肺癌与正常组织的基因表达图谱,我们想从这组数据中寻找差异基因,需要三个文件:原始文件、表型文件、注释文件。
1.原始文件,这里面储存的是每个样本中各个基因的表达量页面底部会提供原始数据,如图所示,点击http下载即可,文件是tar格式,下载下来以后需要解压缩。
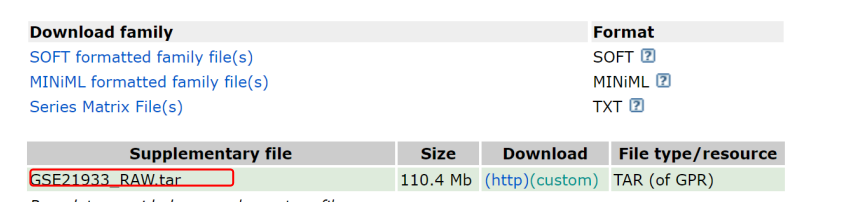
2.表型文件,该文件备注每个样本是属于正常组还是癌症组,想要比较肿瘤和正常样本的区别,我们需要知道每组里面都是样本类型;

这里存储的是样本基因表达量信息;Series Matrix File,(基因表达量矩阵)

3.注释文件,因为原始数据处理得到的差异基因是以探针号表示的,我们需要知道这些探针号代表的基因是什么,这就需要用到注释文件。

有了这三类数据后,就相当于获得了 “烹饪”的原材料,之后便可以对这组数据进行“煎炒炸炖”,依据自己的需求和研究方向进行个性化的数据挖掘。
通常来说,数据挖掘之所以叫“挖掘”是因为需要在海量的数据内,通过检索收集才可以整合出可用信息从而满足我们研究目标。因此这里将介绍一项用于GEO数据下载的利器GEOquery,它是由Davis开发的一款针对下载GEO数据库的R包,目前在R开源网站bio-conductor内,运用它可以简单高效的下载所需的GEO数据。
如何使用GEOquery下载:
if (!require(“BiocManager”, quietly = TRUE))GPT plus 代充 只需 145install.packages("BiocManager")
BiocManager::install(“GEOquery”)
调用:
library(GEOquery)#直接调用 eSet <- getGEO(“GSE21933”,GPT plus 代充 只需 145 destdir = '.', getGPL = F)
getGEO函数会加载GSE的matrix文件,默认会下载其注释探针信息,并对表达矩阵中的探针予以注释,但往往注释文件比较大,会出现parse保存的问题,所以一般建议把注释关掉了:getGPL=F,然后在后续分析步骤里进行手动注释。
我们下载了这些数据,就走完了第一步 “下载数据”,大家如果想了解更多后续个性化分析,请持续关注诺禾致源“GEO数据挖掘”系列文章,每周一个实用干货带您了解上手生信分析。
参考内容
1. https:// bioconductor.org/packag es/release/bioc/html/GEOquery.html
2. https://www. ncbi.nlm.nih.gov/geo/qu ery/acc.cgi?acc=gse21933
各位同学大家好,通过前几篇推文:
生信专栏 |GEO数据库挖掘(一):数据查询与下载+读取
生信专栏 |GEO数据库挖掘(二):下载注释文件+基因symbol ID转换
我们得到了行名是基因名,列名是样本名的表达矩阵,并且已经检查过了是否要log处理,那么在进行差异分析前,我们还有这很关键的一步:探索我们的表达矩阵——是否符合进一步差异分析的要求,话不多说,直接开始:
#得到表达矩阵之后检验一下,测试一下内参基因,画画图,看看是否表达量比较高,如果内参在里面表达很低的话,就要考虑是不是ID转换时出错了 exprSet[‘GAPDH’,] boxplot(exprSet[,1]) #添加样本分组信息 #新建list,根据GEO描述得知前三个为实验组、后3个为对照组 group_list=c(rep(‘hypoxia’,3),rep(‘normal’,3)) group_list #将宽数据变为长数据 library(reshape2)#使用该包中的melt函数 a=rownames(exprSet) a=as.data.frame(a) exprSetplusa=cbind(a,exprSet) # reshape2包是一套重构和整合数据集的万能工具 #需要首先将数据融合(melt),然后将数据重塑(cast)为想要的任何形状 #将多维数据转变为一维数据,即宽数据变为长数据 exp_L = melt(exprSetplusa) head(exp_L) colnames(exp_L)=c(‘symbol’,‘sample’,‘value’) head(exp_L) exp_L$group = rep(group_list,each = nrow(exprSet))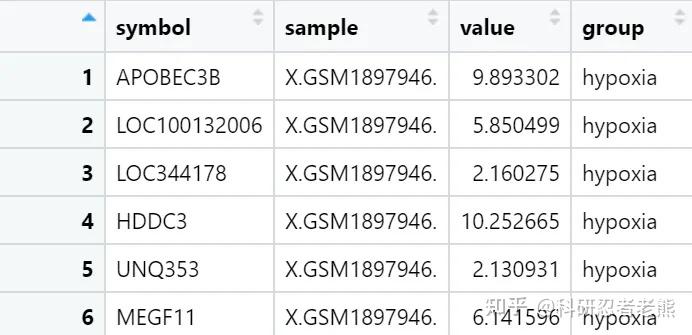
GPT plus 代充 只需 145#画箱线图,看表达矩阵分布图,应该处于差不多水平的才可以进行比较。 #六个样本的表达量中位数应该基本处于同一水平线上 p1 = ggplot(exp_L,aes(x = sample, y = value,fill = group)) +geom_boxplot()+ theme(axis.title.x = element_text(face = ‘italic’),
axis.text.x = element_text(angle = 45 , vjust = 0.5)) print(p1)
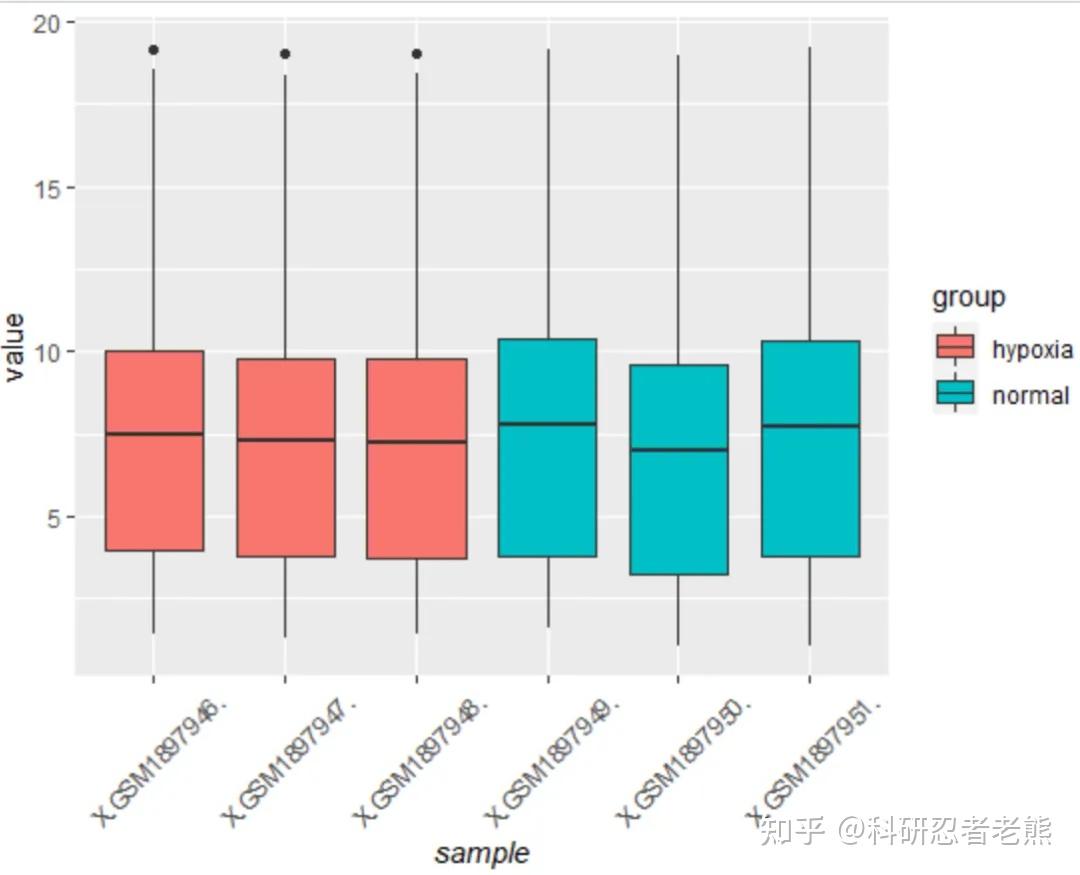
结果又出现大坑,发现这个数据集3vs3的实验设计中,3个样本重复性不好,而理论上他们应该是一样的,这时候就需要用Quntile Normalization方法来校正这个误差,详细的信息在果子老师这篇推文写得很清楚,我贴在这,大家感兴趣的自己去看:
理解 Quntile Normalization
GPT plus 代充 只需 145BiocManager::install(‘preprocessCore’) library(preprocessCore) matrix_exp<-data.matrix(exprSet) class(matrix_exp) nor_exp <- normalize.quantiles(matrix_exp) nor_exp1<-as.data.frame(nor_exp) write.table(nor_exp1, file = “nor_exp1.csv”,sep=“,”, row.names = F,quote = F) #输出之后手动更改一下行和列,然后再把校正过后的数据读取回R中,虽然笨拙,但是非常省力 nor_exp=read.csv(‘nor_exp1.csv’,sep = ‘,’,quote =“”,row.names=1,fill = T,header = T) #校正后重新运行一遍 a=rownames(nor_exp) a=as.data.frame(a) exprSetplusa=cbind(a,nor_exp) exp_L = melt(exprSetplusa) head(exp_L) colnames(exp_L)=c(‘symbol’,‘sample’,‘value’) head(exp_L) exp_L$group = rep(group_list,each = nrow(nor_exp)) #重新画箱线图检验一下 p1 = ggplot(exp_L,aes(x = sample, y = value,fill = group)) +geom_boxplot()+ theme(axis.title.x = element_text(face = ‘italic’),
axis.text.x = element_text(angle = 45 , vjust = 0.5)) print(p1)
这次可以看到数据归一化处理之后变齐了
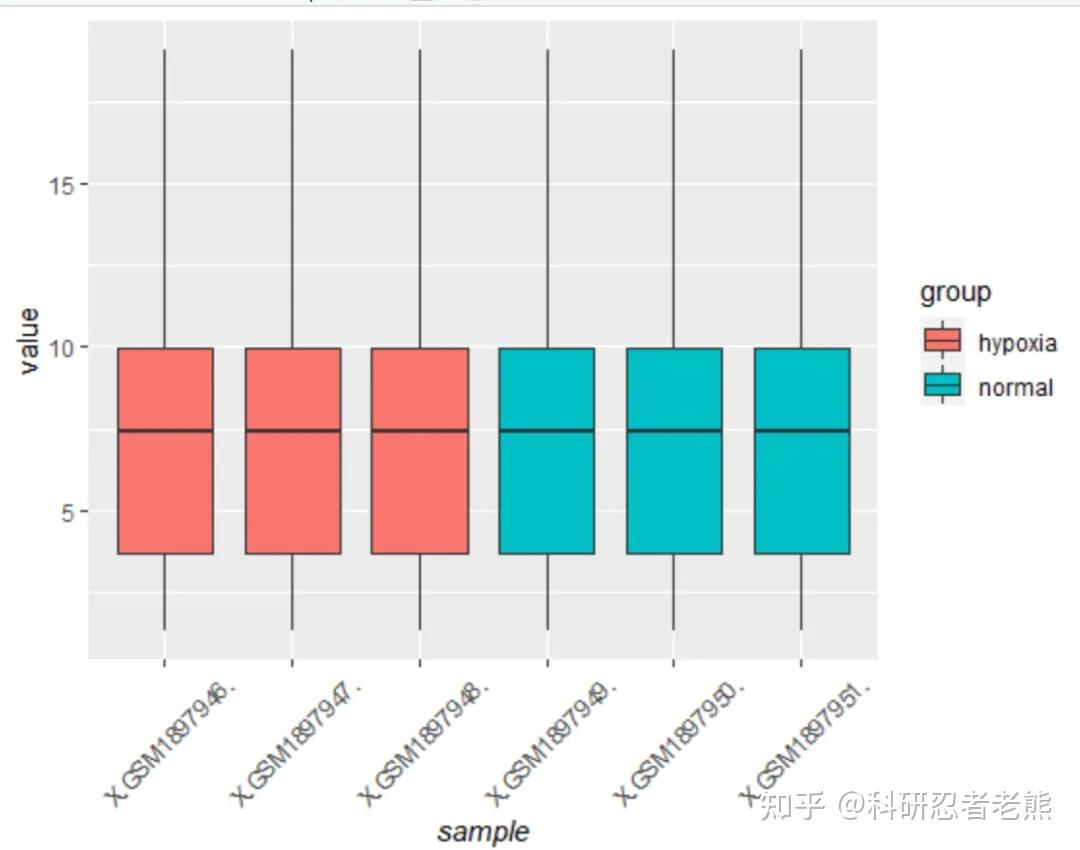
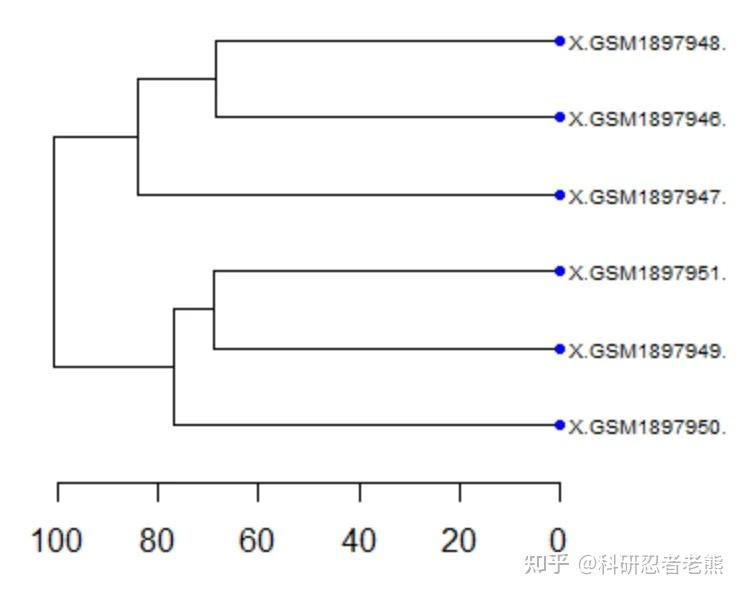
GPT plus 代充 只需 145#聚类分析,相同处理的样本应该是聚在一起的: nodePar <- list(lab.cex = 0.6, pch = c(NA, 19),
cex = 0.7, col = "blue") hc=hclust(dist(t(nor_exp))) par(mar=c(5,5,5,10)) plot(as.dendrogram(hc), nodePar = nodePar, horiz = TRUE)
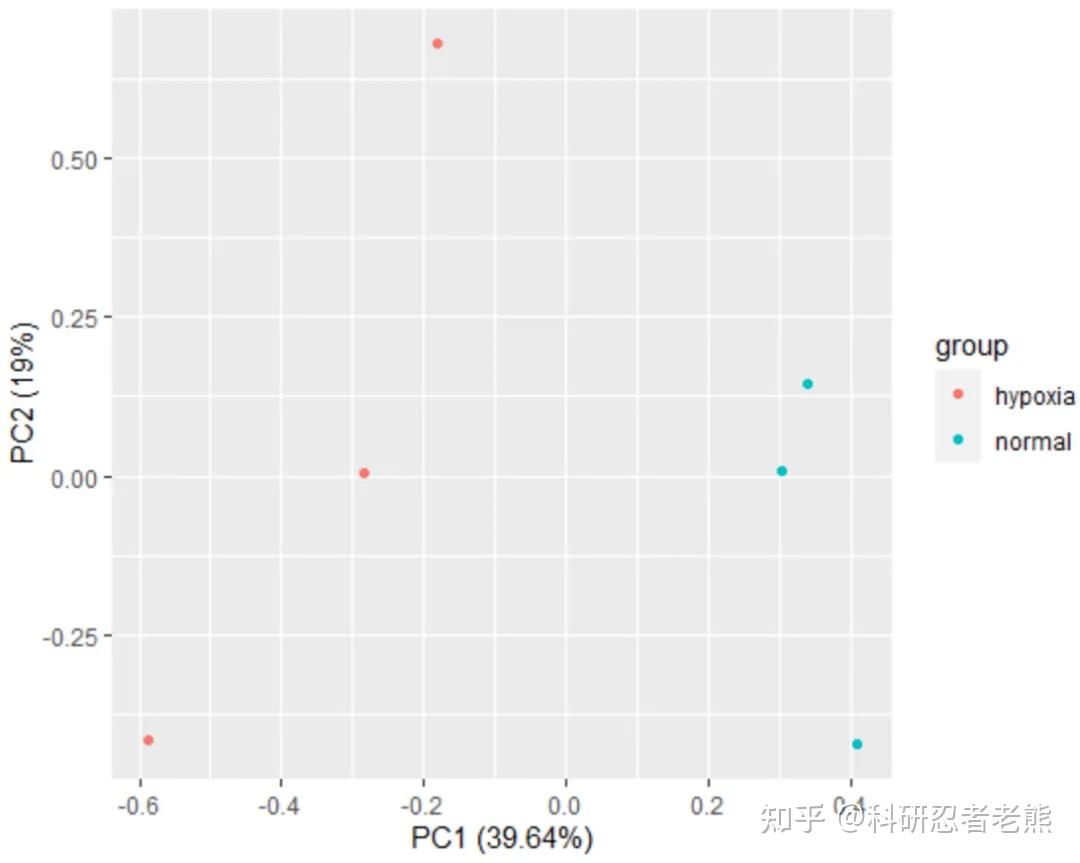
GPT plus 代充 只需 145#主成分分析,相同处理的样本也是聚在一起的: if(! require(“ggfortify”)) install.packages(“ggfortify”) library(ggfortify) df = as.data.frame(t(nor_exp)) df$group = group_list p5 = autoplot(prcomp(df[,1:(ncol(df)-1)]),
data = df,colour = 'group') print(p5)
好了,以上都没问题的话,我们就可以接下来的差异分析啦,我们下一篇就来复现一下差异分析步骤,包括火山图、热图、聚类这些。
别忘了点赞、转发、关注我,让我有动力继续分享!我是老熊,一个立志用通俗易懂的语言带你玩转科研的忍者~
版权声明:本文内容由互联网用户自发贡献,该文观点仅代表作者本人。本站仅提供信息存储空间服务,不拥有所有权,不承担相关法律责任。如发现本站有涉嫌侵权/违法违规的内容,请联系我们,一经查实,本站将立刻删除。
如需转载请保留出处:https://51itzy.com/kjqy/243842.html